Source: European Medicines Agency (EU) Revision Year: 2019 Publisher: Sanofi-aventis groupe, 54, rue La Boétie, F-75008 Paris, France
Pharmacotherapeutic group: Antineoplastic agents, taxanes
ATC code: L01CD04
Cabazitaxel is an antineoplastic agent that acts by disrupting the microtubular network in cells. Cabazitaxel binds to tubulin and promotes the assembly of tubulin into microtubules while simultaneously inhibiting their disassembly. This leads to the stabilisation of microtubules, which results in the inhibition of mitotic and interphase cellular functions.
Cabazitaxel demonstrated a broad spectrum of antitumour activity against advanced human tumours xenografted in mice. Cabazitaxel is active in docetaxel-sensitive tumours. In addition, cabazitaxel demonstrated activity in tumour models insensitive to chemotherapy including docetaxel.
The efficacy and safety of JEVTANA in combination with prednisone or prednisolone were evaluated in a randomised, open-label, international, multi-center, phase III study (EFC6193 study), in patients with metastatic castration resistant prostate cancer previsouly treated with a docetaxel containing regimen.
Overall survival (OS) was the primary efficacy endpoint of the study. Secondary endpoints included Progression Free Survival [PFS (defined as time from randomization to tumour progression, Prostatic Specific Antigen (PSA) progression, pain progression, or death due to any cause, whichever occurred first], Tumour Response Rate based on Response Evaluation Criteria in Solid Tumours (RECIST), PSA Progression (defined as a ≥25% increase or >50% in PSA non-responders or responders respectively), PSA response (declines in serum PSA levels of at least 50%), pain progression [assessed using the Present Pain Intensity (PPI) scale from the McGill-Melzack questionnaire and an Analgesic Score (AS)] and pain response (defined as 2-point greater reduction from baseline median PPI with no concomitant increase in AS, or reduction of ≥50% in analgesic use from baseline mean AS with no concomitant increase in pain).
A total of 755 patients were randomised to receive either JEVTANA 25 mg/m² intravenously every 3 weeks for a maximum of 10 cycles with prednisone or prednisolone 10 mg orally daily (n=378), or to receive mitoxantrone 12 mg/m² intravenously every 3 weeks for a maximum of 10 cycles with prednisone or prednisolone 10 mg orally daily (n=377).
This study included patients over 18 years of age with metastatic castration resistant prostate cancer either measurable by RECIST criteria or non-measurable disease with rising PSA levels or appearance of new lesions, and Eastern Cooperative Oncology Group (ECOG) performance status 0 to 2. Patients had to have neutrophils >1,500/mm³, platelets >100,000/mm³, haemoglobin >10 g/dl, creatinine <1.5 x ULN, total bilirubin <1 x ULN, AST and ALT <1.5 x ULN.
Patients with a history of congestive heart failure, or myocardial infarction within last 6 months, or patients with uncontrolled cardiac arrhythmias, angina pectoris, and/or hypertension were not included in the study.
Demographics, including age, race, and ECOG performance status (0 to 2), were balanced between the treatment arms. In the JEVTANA group, the mean age was 68 years, range (46-92) and the racial distribution was 83.9% Caucasian, 6.9% Asian/Oriental, 5.3% Black and 4% Others.
The median number of cycles was 6 in the JEVTANA group and 4 in the mitoxantrone group. The number of patients who completed the study treatment (10 cycles) was respectively 29.4% and 13.5% in the JEVTANA group and in the comparator group.
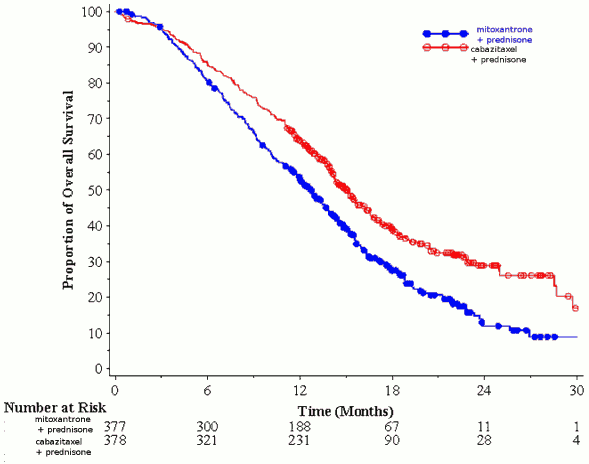
Overall survival was significant longer with JEVTANA compared to mitoxantrone (15.1 months versus 12.7 respectively), with a 30% reduction in the risk of death compared to mitoxantrone (see table 3 and figure 1).
A sub-group of 59 patients received prior cumulative dose of docetaxel <225 mg/m² (29 patients in JEVTANA arm, 30 patients in mitoxantrone arm). There was no significant difference in overall survival in this group of patients (HR (95%CI) 0.96 (0.49-1.86)).
Table 3.Efficacy of JEVTANA in EFC6193 study in the treatment of patients with metastatic castration resistant prostate cancer:
| JEVTANA + prednisone n=378 | mitoxantrone + prednisone n=377 | |
|---|---|---|
| Overall survival | ||
| Number of patients with deaths (%) | 234 (61,9%) | 279 (74%) |
| Median survival (months) (95% CI) | 15,1 (14,1-16,3) | 12,7 (11,6-13,7) |
| Hazard Ratio (HR)1 (95% CI) | 0,70 (0,59-0,83) | |
| p-value | <0,0001 | |
1 HR estimated using Cox model; a hazard ratio of less than 1 favours JEVTANA
Figure 1. Kaplan Meier overall survival curves (EFC6193):
There was an improvement in PFS in the JEVTANA arm compared to mitoxantrone arm, 2.8 (2.4-3.0) months versus 1.4 (1.4-1.7) respectively, HR (95%CI) 0.74 (0.64-0.86), p<0.0001.
There was a significant higher rate of tumour response of 14.4% (95%CI: 9.6-19.3) in patients in the JEVTANA arm compared to 4.4% (95%CI: 1.6-7.2) for patients in the mitoxantrone arm, p=0.0005.
PSA secondary endpoints were positive in the JEVTANA arm. There was a median PSA progression of 6.4 months (95%CI: 5.1-7.3) for patients in JEVTANA arm, compared to 3.1 months (95%CI: 2.2-4.4) in the mitoxantrone arm, HR 0.75 months (95%CI: 0.63-0.90), p=0.0010. The PSA response was 39.2% in patients on JEVTANA arm (95%CI: 33.9-44.5) versus 17.8% of patients on mitoxantrone (95%CI: 13.7-22.0), p=0.0002.
There was no statistical difference between both treatment arms in pain progression and pain response.
In a non-inferiority, multicenter, multinational, randomized, open label phase III study (EFC11785 study), 1200 patients with metastatic castration resistant prostate cancer, previously treated with a docetaxel-containing regimen, were randomized to receive either cabazitaxel 25 mg/m² (n=602) or 20 mg/m² (n=598) dose. Overall survival (OS) was the primary efficacy end-point. The study met its primary objective of demonstrating the non-inferiority of cabazitaxel 20 mg/m² in comparison with 25 mg/m² (see table 4). A statistically significantly higher percentage (p<0.001) of patients showed a PSA response in the 25 mg/m² group (42.9%) compared to the 20 mg/m² group (29.5%). A statistically significantly higher risk of PSA progression in patients with the 20 mg/m² dose with respect to the 25 mg/m² dose was observed (HR 1.195 ; 95%CI: 1.025 to 1.393). There was no statistically difference with regards to the other secondary endpoints (PFS, tumour and pain response, tumour and pain progression, and four subcategories of FACT-P).
Table 4. Overall survival in EFC11785 study in cabazitaxel 25 mg/m² arm versus cabazitaxel 20 mg/m² arm (Intent-to–treat analysis) - Efficacy primary endpoint:
| CBZ20+PRED n=598 | CBZ25+PRED n=602 | |
|---|---|---|
| Overall survival | ||
| Number of deaths, n (%) | 497 (83,1 %) | 501 (83,2%) |
| Median survival (95% CI) (months) | 13,4 (12,19 έως 14,88) | 14,5 (13,47 έως 15,28) |
| Hazard Ratioa | ||
| versus CBZ25+PRED | 1,024 | - |
| 1-sided 98,89% UCI | 1,184 | - |
| 1-sided 95% LCI | 0,922 | - |
CBZ20=Cabazitaxel 20 mg/m², CBZ25=Cabazitaxel 25 mg/m², PRED=Prednisone/Prednisolone
CI=confidence interval, LCI=lower bound of the confidence interval, UCI=upper bound of the confidence interval
a Hazard ratio is estimated using a Cox Proportional Hazards regression model. A hazard ratio <1 indicates a lower risk of cabazitaxel 20 mg/m² with respect to 25 mg/m².
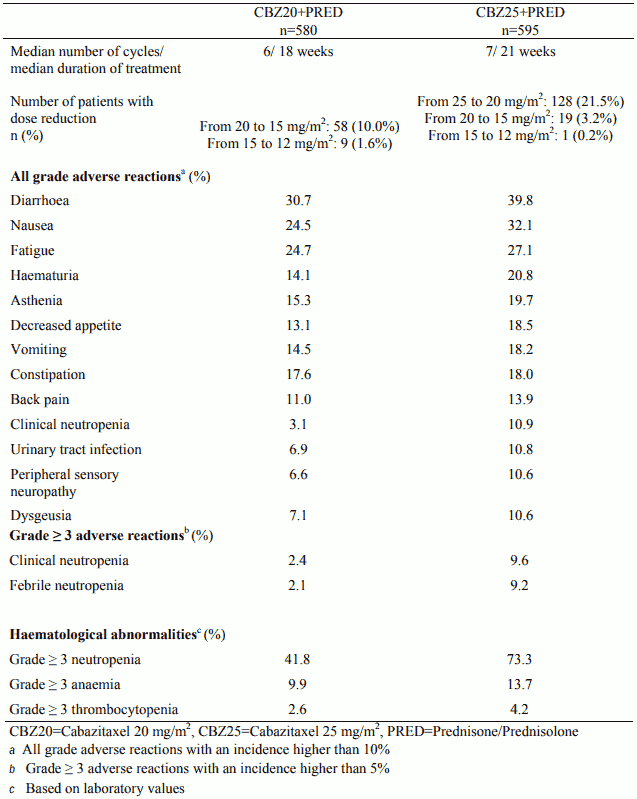
The safety profile of cabazitaxel 25 mg/m² observed in study EFC11785 was qualitatively and quantitatively similar to that observed in the study EFC6193. Study EFC11785 demonstrated a better safety profile for the cabazitaxel 20 mg/m² dose.
Table 5. Summary of safety data for cabazitaxel 25 mg/m² arm versus cabazitaxel 20 mg/m² arm in EFC11785 study:
The European Medicines Agency has waived the obligation to submit the results of studies with JEVTANA in all subsets of the paediatric population in the indication of prostate cancer (see section 4.2 for information on paediatric use).
JEVTANA was evaluated in an open label, multi-center Phase ½ study conducted in a total of 39 paediatric patients (aged between 4 to18 years for the phase 1 part of the study and between 3 to 16 years for the phase 2 part of the study). The phase 2 part did not demonstrate efficacy of cabazitaxel as single agent in paediatric population with recurrent or refractory diffuse intrinsic pontine glioma (DIPG) and high grade glioma (HGG) treated at 30 mg/m².
A population pharmacokinetic analysis was carried out in 170 patients including patients with advanced solid tumours (n=69), metastatic breast cancer (n=34) and metastatic prostate cancer (n=67). These patients received cabazitaxel at doses of 10 to 30 mg/m² weekly or every 3 weeks.
After 1-hour intravenous administration at 25 mg/m² cabazitaxel in patients with metastatic prostate cancer (n=67), the Cmax was 226 ng/ml (Coefficient of Variation (CV): 107%) and was reached at the end of the 1-hour infusion (Tmax). The mean AUC was 991 ng.h/ml (CV: 34%). No major deviation to the dose proportionality was observed from 10 to 30 mg/m² in patients with advanced solid tumours (n=126).
The volume of distribution (Vss) was 4870 l (2640 l/m² for a patient with a median BSA of 1.84 m²) at steady state.
In vitro, the binding of cabazitaxel to human serum proteins was 89-92% and was not saturable up to 50,000 ng/ml, which covers the maximum concentration observed in clinical studies. Cabazitaxel is mainly bound to human serum albumin (82.0%) and lipoproteins (87.9% for HDL, 69.8% for LDL, and 55.8% for VLDL). The in vitro blood-to-plasma concentration ratios in human blood ranged from 0.90 to 0.99 indicating that cabazitaxel was equally distributed between blood and plasma.
Cabazitaxel is extensively metabolised in the liver (>95%), mainly by the CYP3A isoenzyme (80% to 90%). Cabazitaxel is the main circulating compound in human plasma. Seven metabolites were detected in plasma (including 3 active metabolites issued form O-demethylations), with the main one accounting for 5% of parent exposure. Around 20 metabolites of cabazitaxel are excreted into human urine and faeces.
Based on in vitro studies, the potential risk of inhibition by cabazitaxel at clinically relevant concentrations is possible towards medicinal products that are mainly substrate of CYP3A. However a clinical study has shown that cabazitaxel (25 mg/m² administered as a single 1-hour infusion) did not modify the plasma levels of midazolam, a probe substrate of CYP3A. Therefore, at therapeutic doses, co-administration of CYP3A substrates with cabazitaxel to patients is not expected to have any clinical impact.
There is no potential risk of inhibition of medicinal products that are substrates of other CYP enzymes (1A2, 2B6, 2C9, 2C8, 2C19, 2E1, and 2D6) as well as no potential risk of induction by cabazitaxel on medicinal products that are substrates of CYP1A, CYP2C9, and CYP3A. Cabazitaxel did not inhibit in vitro the major biotransformation pathway of warfarin into 7-hydroxywarfarin, which is mediated by CYP2C9. Therefore, no pharmacokinetic interaction of cabazitaxel on warfarin is expected in vivo. In vitro cabazitaxel did not inhibit Multidrug-Resistant Proteins (MRP): MRP1 and MRP2 or Organic Cation Transporter (OCT1). Cabazitaxel inhibited the transport of P-glycoprotein (PgP) (digoxin, vinblastin), Breast-Cancer-Resistant-Proteins (BCRP) (methotrexate) and Organic Anion Transporting Polypeptide OATP1B3 (CCK8) at concentrations at least 15 fold what is observed in clinical setting while it inhibited the transport of OATP1B1 (estradiol-17β-glucuronide) at concentrations only 5 fold what is observed in clinical setting. Therefore the risk of interaction with substrates of MRP, OCT1, PgP, BCRP and OATP1B3 is unlikely in vivo at the dose of 25 mg/m² . The risk of interaction with OATP1B1 transporter is possible, notably during the infusion duration (1 hour) and up to 20 minutes after the end of the infusion (see section 4.5).
After a 1-hour intravenous infusion [14C]-cabazitaxel at 25 mg/m² in patients, approximately 80% of the administered dose was eliminated within 2 weeks. Cabazitaxel is mainly excreted in the faeces as numerous metabolites (76% of the dose); while renal excretion of cabazitaxel and metabolites account for less than 4% of the dose (2.3% as unchanged medicinal product in urine).
Cabazitaxel had a high plasma clearance of 48.5 l/h (26.4 l/h/m² for a patient with a median BSA of 1.84 m²) and a long terminal half-life of 95 hours.
In the population pharmacokinetic analysis in 70 patients of 65 years and older (57 from 65 to 75 and 13 patients above 75), no age effect on the pharmacokinetics of cabazitaxel was observed.
Safety and effectiveness of JEVTANA have not been established in children and adolescents below 18 years of age.
Cabazitaxel is eliminated primarily via liver metabolism.
A dedicated study in 43 cancer patients with hepatic impairment showed no influence of mild (total bilirubin >1 to ≤1.5 x ULN or AST >1.5 x ULN) or moderate (total bilirubin >1.5 to ≤3.0 x ULN) hepatic impairment on cabazitaxel pharmacokinetics. The maximum tolerated dose (MTD) of cabazitaxel was 20 and 15 mg/m², respectively.
In 3 patients with severe hepatic impairment (total bilirubin >3 ULN), a 39% decrease in clearance was observed when compared to patients with mild hepatic impairment, indicating some effect of severe hepatic impairment on cabazitaxel pharmacokinetics. The MTD of cabazitaxel in patients with severe hepatic impairment was not established.
Based on safety and tolerability data, cabazitaxel dose should be reduced in patients with mild hepatic impairment (see sections 4.2, 4.4). JEVTANA is contraindicated in patients with severe hepatic impairment (see section 4.3).
Cabazitaxel is minimally excreted via the kidney (2.3% of the dose). A population pharmacokinetic analysis carried out in 170 patients that included 14 patients with moderate renal impairment (creatinine clearance in the range of 30 to 50 ml/min) and 59 patients with mild renal impairment (creatinine clearance in the range of 50 to 80 ml/min) showed that mild to moderate renal impairment did not have meaningful effects on the pharmacokinetics of cabazitaxel. This was confirmed by a dedicated comparative pharmacokinetic study in solid cancer patients with normal renal function (8 patients), moderate (8 patients) and severe (9 patients) renal impairment, who received several cycles of cabazitaxel in single IV infusion up to 25 mg/m².
Adverse reactions not observed in clinical studies, but seen in dogs after single dose, 5-day and weekly administation at exposure levels lower than clinical exposure levels and with possible relevance to clinical use were arteriolar/periarterolar necrosis in the liver, bile ductule hyperplasia and/or hepatocellular necrosis (see section 4.2).
Adverse reactions not observed in clinical studies, but seen in rats during repeat-dose toxicity studies at exposure levels higher than clinical exposure levels and with possible relevance to clinical use were eye disorders characterized by subcapsular lens fiber swelling/degeneration. These effects were partially reversible after 8 weeks.
Carcinogenicity studies have not been conducted with cabazitaxel. Cabazitaxel did not induce mutations in the bacterial reverse mutation (Ames) test. It was not clastogenic in an in vitro test in human lymphocytes (no induction of structural chromosomal aberration but it increased number of polyploid cells) and induced an increase of micronuclei in the in vivo test in rats. However these genotoxicity findings are inherent to the pharmacological activity of the compound (inhibition of tubulin depolymerization) and have been observed with medicinal products exhibiting the same pharmacological activity.
Cabazitaxel did not affect mating performances or fertility of treated male rats. However, in repeated-dose toxicity studies, degeneration of seminal vesicle and seminiferous tubule atrophy in the testis were observed in rats, and testicular degeneration (minimal epithelial single cell necrosis in epididymis), was observed in dogs. Exposures in animals were similar or lower than those seen in humans receiving clinically relevant doses of cabazitaxel.
Cabazitaxel induced embryofoetal toxicity in female rats treated intravenously once daily from gestational days 6 through 17 linked with maternal toxicity and consisted of foetal deaths and decreased mean foetal weight associated with delay in skeletal ossification. Exposures in animals were lower than those seen in humans receiving clinically relevant doses of cabazitaxel. Cabazitaxel crossed the placenta barrier in rats.
In rats, cabazitaxel and its metabolites are excreted in maternal milk at a quantity up to 1.5% of administered dose over 24 hours.
Results of environmental risk assessment studies indicated that use of JEVTANA will not cause significant risk to the aquatic environment (see section 6.6 for disposal of unused product).
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.