Source: European Medicines Agency (EU) Revision Year: 2019 Publisher: Novartis Europharm Limited, Vista Building, Elm Park, Merrion Road, Dublin 4, Ireland
Hypersensitivity to the active substance or to any of the excipients listed in section 6.1.
Combination with other iron chelator therapies as the safety of such combinations has not been established (see section 4.5).
Patients with estimated creatinine clearance <60 ml/min.
Deferasirox has been studied only in patients with baseline serum creatinine within the age-appropriate normal range.
During clinical studies, increases in serum creatinine of >33% on ≥2 consecutive occasions, sometimes above the upper limit of the normal range, occurred in about 36% of patients. These were dose-dependent. About two-thirds of the patients showing serum creatinine increase returned below the 33% level without dose adjustment. In the remaining third the serum creatinine increase did not always respond to a dose reduction or a dose interruption. In some cases, only a stabilisation of the serum creatinine values has been observed after dose reduction. Cases of acute renal failure have been reported following post-marketing use of deferasirox (see section 4.8). In some post-marketing cases, renal function deterioration has led to renal failure requiring temporary or permanent dialysis.
The causes of the rises in serum creatinine have not been elucidated. Particular attention should therefore be paid to monitoring of serum creatinine in patients who are concomitantly receiving medicinal products that depress renal function, and in patients who are receiving high doses of deferasirox and/or low rates of transfusion (<7 ml/kg/month of packed red blood cells or <2 units/month for an adult). While no increase in renal adverse events was observed after dose escalation of EXJADE dispersible tablets to doses above 30 mg/kg in clinical studies, an increased risk of renal adverse events with EXJADE dispersible tablet doses above 30 mg/kg cannot be excluded.
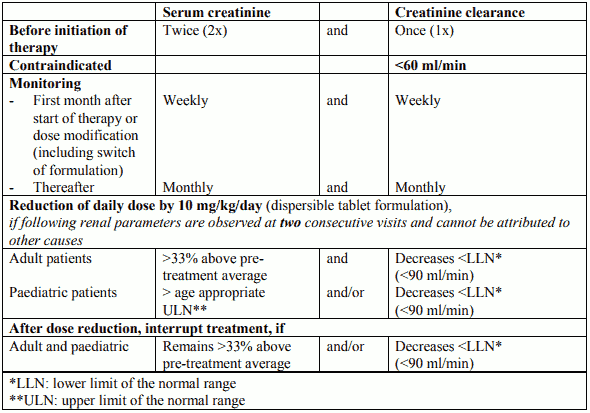
It is recommended that serum creatinine be assessed in duplicate before initiating therapy. Serum creatinine, creatinine clearance (estimated with the Cockcroft-Gault or MDRD formula in adults and with the Schwartz formula in children) and/or plasma cystatin C levels should be monitored prior to therapy, weekly in the first month after initiation or modification of therapy with EXJADE (including switch of formulation), and monthly thereafter. Patients with pre-existing renal conditions and patients who are receiving medicinal products that depress renal function may be more at risk of complications. Care should be taken to maintain adequate hydration in patients who develop diarrhoea or vomiting.
There have been post-marketing reports of metabolic acidosis occurring during treatment with deferasirox. The majority of these patients had renal impairment, renal tubulopathy (Fanconi syndrome) or diarrhoea, or conditions where acid-base imbalance is a known complication. Acid-base balance should be monitored as clinically indicated in these populations. Interruption of EXJADE therapy should be considered in patients who develop metabolic acidosis.
Post-marketing cases of severe forms of renal tubulopathy (such as Fanconi syndrome) and renal failure associated with changes in consciousness in the context of hyperammonaemic encephalopathy have been reported in patients treated with deferasirox, mainly in children. It is recommended that hyperammonaemic encephalopathy be considered and ammonia levels measured in patients who develop unexplained changes in mental status while on Exjade therapy.
Table 3. Dose adjustment and interruption of treatment for renal monitoring
Treatment may be reinitiated depending on the individual clinical circumstances.
Dose reduction or interruption may be also considered if abnormalities occur in levels of markers of renal tubular function and/or as clinically indicated:
Renal tubulopathy has been mainly reported in children and adolescents with beta-thalassaemia treated with EXJADE.
Patients should be referred to a renal specialist, and further specialised investigations (such as renal biopsy) may be considered if the following occur despite dose reduction and interruption:
Liver function test elevations have been observed in patients treated with deferasirox. Post-marketing cases of hepatic failure, some of which were fatal, have been reported. Severe forms associated with changes in consciousness in the context of hyperammonaemic encephalopathy, may occur in patients treated with deferasirox, particularly in children. It is recommended that hyperammonaemic encephalopathy be considered and ammonia levels measured in patients who develop unexplained changes in mental status while on Exjade therapy. Care should be taken to maintain adequate hydration in patients who experience volume-depleting events (such as diarrhoea or vomiting), particularly in children with acute illness. Most reports of hepatic failure involved patients with significant morbidities including pre-existing liver cirrhosis. However, the role of deferasirox as a contributing or aggravating factor cannot be excluded (see section 4.8).
It is recommended that serum transaminases, bilirubin and alkaline phosphatase be checked before the initiation of treatment, every 2 weeks during the first month and monthly thereafter. If there is a persistent and progressive increase in serum transaminase levels that cannot be attributed to other causes, EXJADE should be interrupted. Once the cause of the liver function test abnormalities has been clarified or after return to normal levels, cautious re-initiation of treatment at a lower dose followed by gradual dose escalation may be considered.
EXJADE is not recommended in patients with severe hepatic impairment (Child-Pugh Class C) (see section 5.2).
Table 4. Summary of safety monitoring recommendations
| Test | Frequency |
|---|---|
| Serum creatinine | In duplicate prior to therapy. Weekly during first month of therapy or after dose modification (including switch of formulation). Monthly thereafter. |
| Creatinine clearance and/or plasma cystatin C | Prior to therapy. Weekly during first month of therapy or after dose modification (including switch of formulation). Monthly thereafter. |
| Proteinuria | Prior to therapy. Monthly thereafter. |
| Other markers of renal tubular function (such as glycosuria in non-diabetics and low levels of serum potassium, phosphate, magnesium or urate, phosphaturia, aminoaciduria) | As needed. |
| Serum transaminases, bilirubin, alkaline phosphatase | Prior to therapy. Every 2 weeks during first month of therapy. Monthly thereafter. |
| Auditory and ophthalmic testing | Prior to therapy. Annually thereafter. |
| Body weight, height and sexual development | Prior to therapy. Annually in paediatric patients. |
Test Frequency Serum creatinine In duplicate prior to therapy. Weekly during first month of therapy or after dose modification (including switch of formulation). Monthly thereafter. Creatinine clearance and/or plasma cystatin C Prior to therapy. Weekly during first month of therapy or after dose modification (including switch of formulation). Monthly thereafter. Proteinuria Prior to therapy. Monthly thereafter. Other markers of renal tubular function (such as glycosuria in non-diabetics and low levels of serum potassium, phosphate, magnesium or urate, phosphaturia, aminoaciduria) As needed. Serum transaminases, bilirubin, alkaline phosphatase Prior to therapy. Every 2 weeks during first month of therapy. Monthly thereafter. Auditory and ophthalmic testing Prior to therapy. Annually thereafter. Body weight, height and sexual development Prior to therapy. Annually in paediatric patients.
In patients with a short life expectancy (e.g. high-risk myelodysplastic syndromes), especially when co-morbidities could increase the risk of adverse events, the benefit of EXJADE might be limited and may be inferior to risks. As a consequence, treatment with EXJADE is not recommended in these patients.
Caution should be used in elderly patients due to a higher frequency of adverse reactions (in particular, diarrhoea).
Data in children with non-transfusion-dependent thalassaemia are very limited (see section 5.1). As a consequence, EXJADE therapy should be closely monitored to detect adverse reactions and to follow iron burden in the paediatric population. In addition, before treating heavily iron-overloaded children with non-transfusion-dependent thalassaemia with EXJADE, the physician should be aware that the consequences of long-term exposure in such patients are currently not known.
Upper gastrointestinal ulceration and haemorrhage have been reported in patients, including children and adolescents, receiving deferasirox. Multiple ulcers have been observed in some patients (see section 4.8). There have been reports of ulcers complicated with digestive perforation. Also, there have been reports of fatal gastrointestinal haemorrhages, especially in elderly patients who had haematological malignancies and/or low platelet counts. Physicians and patients should remain alert for signs and symptoms of gastrointestinal ulceration and haemorrhage during EXJADE therapy and promptly initiate additional evaluation and treatment if a serious gastrointestinal adverse reaction is suspected. Caution should be exercised in patients who are taking EXJADE in combination with substances that have known ulcerogenic potential, such as NSAIDs, corticosteroids, or oral bisphosphonates, in patients receiving anticoagulants and in patients with platelet counts below 50,000/mm³ (50 × 109/l) (see section 4.5).
Skin rashes may appear during EXJADE treatment. The rashes resolve spontaneously in most cases. When interruption of treatment may be necessary, treatment may be reintroduced after resolution of the rash, at a lower dose followed by gradual dose escalation. In severe cases this reintroduction could be conducted in combination with a short period of oral steroid administration. Severe cutaneous adverse reactions (SCARs) including Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN) and drug reaction with eosinophilia and systemic symptoms (DRESS), which could be life-threatening or fatal, have been reported. If any SCAR is suspected, EXJADE should be discontinued immediately and should not be reintroduced. At the time of prescription, patients should be advised of the signs and symptoms of severe skin reactions, and be closely monitored.
Cases of serious hypersensitivity reactions (such as anaphylaxis and angioedema) have been reported in patients receiving deferasirox, with the onset of the reaction occurring in the majority of cases within the first month of treatment (see section 4.8). If such reactions occur, EXJADE should be discontinued and appropriate medical intervention instituted. Deferasirox should not be reintroduced in patients who have experienced a hypersensitivity reaction due to the risk of anaphylactic shock (see section 4.3).
Auditory (decreased hearing) and ocular (lens opacities) disturbances have been reported (see section 4.8). Auditory and ophthalmic testing (including fundoscopy) is recommended before the start of treatment and at regular intervals thereafter (every 12 months). If disturbances are noted during the treatment, dose reduction or interruption may be considered.
There have been post-marketing reports of leukopenia, thrombocytopenia or pancytopenia (or aggravation of these cytopenias) and of aggravated anaemia in patients treated with deferasirox. Most of these patients had pre-existing haematological disorders that are frequently associated with bone marrow failure. However, a contributory or aggravating role cannot be excluded. Interruption of treatment should be considered in patients who develop unexplained cytopenia.
Monthly monitoring of serum ferritin is recommended in order to assess the patient’s response to therapy and to avoid overchelation (see section 4.2). Dose reduction or closer monitoring of renal and hepatic function, and serum ferritin levels are recommended during periods of treatment with high doses and when serum ferritin levels are close to the target range. If serum ferritin falls consistently below 500 µg/l (in transfusional iron overload) or below 300 µg/l (in non-transfusion-dependent thalassaemia syndromes), an interruption of treatment should be considered.
The results of the tests for serum creatinine, serum ferritin and serum transaminases should be recorded and regularly assessed for trends.
In two clinical studies, growth and sexual development of paediatric patients treated with deferasirox for up to 5 years were not affected (see section 4.8). However, as a general precautionary measure in the management of paediatric patients with transfusional iron overload, body weight, height and sexual development should be monitored prior to therapy and at regular intervals (every 12 months).
Cardiac dysfunction is a known complication of severe iron overload. Cardiac function should be monitored in patients with severe iron overload during long-term treatment with EXJADE.
The dispersible tablets contain lactose.
Patients with rare hereditary problems of galactose intolerance, the Lapp lactase deficiency or glucose-galactose malabsorption should not take this medicinal product.
The safety of deferasirox in combination with other iron chelators has not been established. Therefore, it must not be combined with other iron chelator therapies (see section 4.3).
The bioavailability of deferasirox was increased to a variable extent when taken along with food. EXJADE dispersible tablets must therefore be taken on an empty stomach at least 30 minutes before food, preferably at the same time each day (see sections 4.2 and 5.2).
Deferasirox metabolism depends on UGT enzymes. In a healthy volunteer study, the concomitant administration of deferasirox (single dose of 30 mg/kg, dispersible tablet formulation) and the potent UGT inducer, rifampicin, (repeated dose of 600 mg/day) resulted in a decrease of deferasirox exposure by 44% (90% CI: 37%-51%). Therefore, the concomitant use of EXJADE with potent UGT inducers (e.g. rifampicin, carbamazepine, phenytoin, phenobarbital, ritonavir) may result in a decrease in EXJADE efficacy. The patient’s serum ferritin should be monitored during and after the combination, and the dose of EXJADE adjusted if necessary.
Cholestyramine significantly reduced the deferasirox exposure in a mechanistic study to determine the degree of enterohepatic recycling (see section 5.2).
In a healthy volunteer study, the concomitant administration of deferasirox dispersible tablets and midazolam (a CYP3A4 probe substrate) resulted in a decrease of midazolam exposure by 17% (90% CI: 8%-26%). In the clinical setting, this effect may be more pronounced. Therefore, due to a possible decrease in efficacy, caution should be exercised when deferasirox is combined with substances metabolised through CYP3A4 (e.g. ciclosporin, simvastatin, hormonal contraceptive agents, bepridil, ergotamine).
In a healthy volunteer study, the concomitant administration of deferasirox as a moderate CYP2C8 inhibitor (30 mg/kg daily, dispersible tablet formulation), with repaglinide, a CYP2C8 substrate, given as a single dose of 0.5 mg, increased repaglinide AUC and Cmax about 2.3-fold (90% CI [2.03-2.63]) and 1.6-fold (90% CI [1.42-1.84]), respectively. Since the interaction has not been established with dosages higher than 0.5 mg for repaglinide, the concomitant use of deferasirox with repaglinide should be avoided. If the combination appears necessary, careful clinical and blood glucose monitoring should be performed (see section 4.4). An interaction between deferasirox and other CYP2C8 substrates like paclitaxel cannot be excluded.
In a healthy volunteer study, the concomitant administration of deferasirox as a CYP1A2 inhibitor (repeated dose of 30 mg/kg/day, dispersible tablet formulation) and the CYP1A2 substrate theophylline (single dose of 120 mg) resulted in an increase of theophylline AUC by 84% (90% CI: 73% to 95%). The single dose Cmax was not affected, but an increase of theophylline Cmax is expected to occur with chronic dosing. Therefore, the concomitant use of deferasirox with theophylline is not recommended. If deferasirox and theophylline are used concomitantly, monitoring of theophylline concentration and theophylline dose reduction should be considered. An interaction between deferasirox and other CYP1A2 substrates cannot be excluded. For substances that are predominantly metabolised by CYP1A2 and that have a narrow therapeutic index (e.g. clozapine, tizanidine), the same recommendations apply as for theophylline.
The concomitant administration of deferasirox and aluminium-containing antacid preparations has not been formally studied. Although deferasirox has a lower affinity for aluminium than for iron, it is not recommended to take deferasirox tablets with aluminium-containing antacid preparations.
The concomitant administration of deferasirox with substances that have known ulcerogenic potential, such as NSAIDs (including acetylsalicylic acid at high dosage), corticosteroids or oral bisphosphonates may increase the risk of gastrointestinal toxicity (see section 4.4). The concomitant administration of deferasirox with anticoagulants may also increase the risk of gastrointestinal haemorrhage. Close clinical monitoring is required when deferasirox is combined with these substances.
Concomitant administration of deferasirox and busulfan resulted in an increase of busulfan exposure (AUC), but the mechanism of the interaction remains unclear. If possible, evaluation of the pharmacokinetics (AUC, clearance) of a busulfan test dose should be performed to allow dose adjustment.
No clinical data on exposed pregnancies are available for deferasirox. Studies in animals have shown some reproductive toxicity at maternally toxic doses (see section 5.3). The potential risk for humans is unknown.
As a precaution, it is recommended that EXJADE is not used during pregnancy unless clearly necessary.
EXJADE may decrease the efficacy of hormonal contraceptives (see section 4.5). Women of childbearing potential are recommended to use additional or alternative non-hormonal methods of contraception when using EXJADE.
In animal studies, deferasirox was found to be rapidly and extensively secreted into maternal milk. No effect on the offspring was noted. It is not known if deferasirox is secreted into human milk. Breast-feeding while taking EXJADE is not recommended.
No fertility data is available for humans. In animals, no adverse effects on male or female fertility were found (see section 5.3).
EXJADE has minor influence on the ability to drive and use machines. Patients experiencing the uncommon adverse reaction of dizziness should exercise caution when driving or operating machines (see section 4.8).
The most frequent reactions reported during chronic treatment with deferasirox dispersible tablets in adult and paediatric patients include gastrointestinal disturbances (mainly nausea, vomiting, diarrhoea or abdominal pain) and skin rash. Diarrhoea is reported more commonly in paediatric patients aged 2 to 5 years and in the elderly. These reactions are dose-dependent, mostly mild to moderate, generally transient and mostly resolve even if treatment is continued.
During clinical studies, dose-dependent increases in serum creatinine occurred in about 36% of patients, though most remained within the normal range. Decreases in mean creatinine clearance have been observed in both paediatric and adult patients with beta-thalassemia and iron overload during the first year of treatment, but there is evidence that this does not decrease further in subsequent years of treatment. Elevations of liver transaminases have been reported. Safety monitoring schedules for renal and liver parameters are recommended. Auditory (decreased hearing) and ocular (lens opacities) disturbances are uncommon, and yearly examinations are also recommended (see section 4.4).
Severe cutaneous adverse reactions (SCARs), including Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN) and drug reaction with eosinophilia and systemic symptoms (DRESS) have been reported with the use of EXJADE (see section 4.4).
Adverse reactions are ranked below using the following convention: very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1,000 to <1/100); rare (≥1/10,000 to <1/1,000); very rare (<1/10,000); not known (cannot be estimated from the available data). Within each frequency grouping, adverse reactions are presented in order of decreasing seriousness.
Table 5
Not known: Pancytopenia1, thrombocytopenia1, anaemia aggravated1, neutropenia1
Not known: Hypersensitivity reactions (including anaphylactic reactions and angioedema)1
Not known: Metabolic acidosis1
Uncommon: Anxiety, sleep disorder
Common: Headache
Uncommon: Dizziness
Uncommon: Cataract, maculopathy
Rare: Optic neuritis
Uncommon: Deafness
Uncommon: Laryngeal pain
Common: Diarrhoea, constipation, vomiting, nausea, abdominal pain, abdominal distension, dyspepsia
Uncommon: Gastrointestinal haemorrhage, gastric ulcer (including multiple ulcers), duodenal ulcer, gastritis
Rare: Oesophagitis
Not known: Gastrointestinal perforation1, acute pancreatitis1
Common: Transaminases increased
Uncommon: Hepatitis, cholelithiasis
Not known: Hepatic failure1,2
Common: Rash, pruritus
Uncommon: Pigmentation disorder
Rare: Drug reaction with eosinophilia and systemic symptoms (DRESS)
Not known: Stevens-Johnson syndrome1, hypersensitivity vasculitis1, urticaria1, erythema multiforme1, alopecia1, toxic epidermal necrolysis (TEN)1
Very common: Blood creatinine increased
Common: Proteinuria
Uncommon: Renal tubular disorder2 (acquired Fanconi syndrome), glycosuria
Not known: Acute renal failure1,2, tubulointerstitial nephritis1, nephrolithiasis1, renal tubular necrosis1
Uncommon: Pyrexia, oedema, fatigue
1 Adverse reactions reported during post-marketing experience. These are derived from spontaneous reports for which it is not always possible to reliably establish frequency or a causal relationship to exposure to the medicinal product.
2 Severe forms associated with changes in consciousness in the context of hyperammonaemic encephalopathy have been reported.
Gallstones and related biliary disorders were reported in about 2% of patients. Elevations of liver transaminases were reported as an adverse reaction in 2% of patients. Elevations of transaminases greater than 10 times the upper limit of the normal range, suggestive of hepatitis, were uncommon (0.3%). During post-marketing experience, hepatic failure, sometimes fatal, has been reported with the deferasirox dispersible tablet formulation, especially in patients with pre-existing liver cirrhosis (see section 4.4). There have been post-marketing reports of metabolic acidosis. The majority of these patients had renal impairment, renal tubulopathy (Fanconi syndrome) or diarrhoea, or conditions where acid-base imbalance is a known complication (see section 4.4). Cases of serious acute pancreatitis were observed without documented underlying biliary conditions. As with other iron chelator treatment, high-frequency hearing loss and lenticular opacities (early cataracts) have been uncommonly observed in patients treated with deferasirox (see section 4.4).
In a retrospective meta-analysis of 2,102 adult and paediatric beta-thalassaemia patients with transfusional iron overload treated with deferasirox dispersible tablets in two randomised and four open label studies of up to five years' duration, a mean creatinine clearance decrease of 13.2% in adult patients (95% CI: -14.4% to -12.1%; n=935) and 9.9% (95% CI: -11.1% to -8.6%; n=1,142) in paediatric patients was observed during the first year of treatment. In 250 patients who were followed for up to five years, no further decrease in mean creatinine clearance levels was observed.
In a 1-year study in patients with non-transfusion-dependent thalassaemia syndromes and iron overload (dispersible tablets at a dose of 10 mg/kg/day), diarrhoea (9.1%), rash (9.1%), and nausea (7.3%) were the most frequent study drug-related adverse events. Abnormal serum creatinine and creatinine clearance values were reported in 5.5% and 1.8% of patients, respectively. Elevations of liver transaminases greater than 2 times the baseline and 5 times the upper limit of normal were reported in 1.8% of patients.
In two clinical studies, growth and sexual development of paediatric patients treated with deferasirox for up to 5 years were not affected (see section 4.4).
Diarrhoea is reported more commonly in paediatric patients aged 2 to 5 years than in older patients.
Renal tubulopathy has been mainly reported in children and adolescents with beta-thalassaemia treated with deferasirox. In post-marketing reports, a high proportion of cases of metabolic acidosis occurred in children in the context of Fanconi syndrome.
Acute pancreatitis has been reported, particularly in children and adolescents.
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed in Appendix V.
Dispersion in carbonated drinks or milk is not recommended due to foaming and slow dispersion, respectively.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.