AMERGE Film-coated tablet Ref.[10746] Active ingredients: Naratriptan
Source: FDA, National Drug Code (US) Revision Year: 2020
12.1. Mechanism of Action
Naratriptan binds with high affinity to human cloned 5-HT1B/1D receptors. Migraines are likely due to local cranial vasodilatation and/or to the release of sensory neuropeptides (including substance P and calcitonin gene-related peptide) through nerve endings in the trigeminal system. The therapeutic activity of AMERGE for the treatment of migraine headache is thought to be due to the agonist effects at the 5-HT1B/1D receptors on intracranial blood vessels (including the arterio-venous anastomoses) and sensory nerves of the trigeminal system, which result in cranial vessel constriction and inhibition of pro-inflammatory neuropeptide release.
12.2. Pharmacodynamics
In the anesthetized dog, naratriptan has been shown to reduce the carotid arterial blood flow with little or no effect on arterial blood pressure or total peripheral resistance. While the effect on blood flow was selective for the carotid arterial bed, increases in vascular resistance of up to 30% were seen in the coronary arterial bed. Naratriptan has also been shown to inhibit trigeminal nerve activity in rat and cat.
In 10 subjects with suspected CAD undergoing coronary artery catheterization, there was a 1% to 10% reduction in coronary artery diameter following subcutaneous injection of 1.5 mg of naratriptan [see Contraindications (4)].
12.3. Pharmacokinetics
Absorption
Naratriptan is well absorbed, with about 70% oral bioavailability. Following administration of a 2.5-mg tablet, the peak concentrations are obtained in 2 to 3 hours. After administration of 1- or 2.5-mg tablets, the Cmax is somewhat (about 50%) higher in women (not corrected for milligram-per-kilogram dose) than in men. During a migraine attack, absorption is slower, with a Tmax of 3 to 4 hours. Food does not affect the pharmacokinetics of naratriptan. Naratriptan displays linear kinetics over the therapeutic dose range.
Distribution
The steady-state volume of distribution of naratriptan is 170 L. Plasma protein binding is 28% to 31% over the concentration range of 50 to 1,000 ng/mL.
Metabolism
In vitro, naratriptan is metabolized by a wide range of cytochrome P450 isoenzymes into a number of inactive metabolites.
Elimination
Naratriptan is predominantly eliminated in urine, with 50% of the dose recovered unchanged and 30% as metabolites in urine. The mean elimination half-life of naratriptan is 6 hours. The systemic clearance of naratriptan is 6.6 mL/min/kg. The renal clearance (220 mL/min) exceeds glomerular filtration rate, indicating active tubular secretion. Repeat administration of naratriptan tablets does not result in drug accumulation.
Special Populations
Age
A small decrease in clearance (approximately 26%) was observed in healthy elderly subjects (65 to 77 years) compared with younger subjects, resulting in slightly higher exposure [see Use in Specific Populations (8.5)].
Race
The effect of race on the pharmacokinetics of naratriptan has not been examined.
Renal Impairment
Clearance of naratriptan was reduced by 50% in subjects with moderate renal impairment (creatinine clearance: 18 to 39 mL/min) compared with the normal group. Decrease in clearances resulted in an increase of mean half-life from 6 hours (healthy) to 11 hours (range: 7 to 20 hours). The mean Cmax increased by approximately 40%. The effects of severe renal impairment (creatinine clearance: ≤15 mL/min) on the pharmacokinetics of naratriptan have not been assessed [see Contraindications (4)].
Hepatic Impairment
Clearance of naratriptan was decreased by 30% in subjects with moderate hepatic impairment (Child-Pugh Grade A or B). This resulted in an approximately 40% increase in the half-life (range: 8 to 16 hours). The effects of severe hepatic impairment (Child-Pugh Grade C) on the pharmacokinetics of naratriptan have not been assessed [see Contraindications (4)].
Drug Interaction Studies
From population pharmacokinetic analyses, coadministration of naratriptan and fluoxetine, beta-blockers, or tricyclic antidepressants did not affect the clearance of naratriptan.
Oral Contraceptives
Oral contraceptives reduced clearance by 32% and volume of distribution by 22%, resulting in slightly higher concentrations of naratriptan. Hormone replacement therapy had no effect on pharmacokinetics in older female patients.
Monoamine Oxidase and P450 Inhibitors
Naratriptan does not inhibit monoamine oxidase (MAO) enzymes and is a poor inhibitor of P450; metabolic interactions between naratriptan and drugs metabolized by P450 or MAO are therefore unlikely.
Smoking
Smoking increased the clearance of naratriptan by 30%.
Alcohol
In normal volunteers, co-administration of single doses of naratriptan tablets and alcohol did not result in substantial modification of naratriptan pharmacokinetic parameters.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
In carcinogenicity studies, mice and rats were given naratriptan by oral gavage for 104 weeks. There was no evidence of an increase in tumors related to naratriptan administration in mice receiving up to 200 mg/kg/day. That dose was associated with a plasma (AUC) exposure that was 110 times the exposure in humans receiving the MRDD of 5 mg. Two rat studies were conducted, one using a standard diet and the other a nitrite-supplemented diet (naratriptan can be nitrosated in vitro to form a mutagenic product that has been detected in the stomachs of rats fed a high-nitrite diet). Doses of 5, 20, and 90 mg/kg were associated with AUC exposures that in the standard-diet study were 7, 40, and 236 times, respectively, and in the nitrite-supplemented–diet study were 7, 29, and 180 times, respectively, the exposure in humans at the MRDD. In both studies, there was an increase in the incidence of thyroid follicular hyperplasia in high-dose males and females and in thyroid follicular adenomas in high-dose males. In the standard-diet study only, there was also an increase in the incidence of benign c-cell adenomas in the thyroid of high-dose males and females. The exposures achieved at the no-effect dose for thyroid tumors were 40 (standard diet) and 29 (nitrite-supplemented diet) times the exposure achieved in humans at the MRDD. In the nitrite-supplemented–diet study only, the incidence of benign lymphocytic thymoma was increased in all treated groups of females. It was not determined if the nitrosated product is systemically absorbed. However, no changes were seen in the stomachs of rats in that study.
Mutagenesis
Naratriptan was not mutagenic when tested in in vitro gene mutation (Ames and mouse lymphoma tk) assays. Naratriptan was also negative in the in vitro human lymphocyte assay and the in vivo mouse micronucleus assay. Naratriptan can be nitrosated in vitro to form a mutagenic product (WHO nitrosation assay) that has been detected in the stomachs of rats fed a nitrite-supplemented diet.
Impairment of Fertility
In a reproductive toxicity study in which male and female rats were administered naratriptan orally prior to and throughout the mating period (10, 60, 170, or 340 mg/kg/day; plasma exposures [AUC] approximately 11, 70, 230, and 470 times, respectively, the human exposure at the MRDD), there was a drug-related decrease in the number of females exhibiting normal estrous cycles at doses of 170 mg/kg/day or greater and an increase in pre-implantation loss at 60 mg/kg/day or greater. In high-dose males, testicular/epididymal atrophy accompanied by spermatozoa depletion reduced mating success and may have contributed to the observed pre-implantation loss. The exposures achieved at the no-effect doses for pre-implantation loss, anestrus, and testicular effects were approximately 11, 70, and 230 times, respectively, the exposures in humans at the MRDD.
In a study in which rats were dosed orally with naratriptan (10, 60, or 340 mg/kg/day) for 6 months, changes in the female reproductive tract including atrophic or cystic ovaries and anestrus were seen at the high dose. The exposure at the no-effect dose of 60 mg/kg was approximately 85 times that in humans at the MRDD.
14. Clinical Studies
The efficacy of AMERGE in the acute treatment of migraine headaches was evaluated in 3 randomized, double-blind, placebo-controlled trials in adult patients (Trials 1, 2, 3). These trials enrolled adult patients who were predominantly female (86%) and Caucasian (96%) with a mean age of 41 years (range: 18 to 65 years). In all studies, patients were instructed to treat at least 1 moderate to severe headache. Headache response, defined as a reduction in headache severity from moderate or severe pain to mild or no pain, was assessed up to 4 hours after dosing. Associated symptoms such as nausea, vomiting, photophobia, and phonophobia were also assessed. Maintenance of response was assessed for up to 24 hours postdose. A second dose of AMERGE or other rescue medication to treat migraines was allowed 4 to 24 hours after the initial treatment for recurrent headache.
In all 3 trials, the percentage of patients achieving headache response 4 hours after treatment, the primary outcome measure, was significantly greater among patients receiving AMERGE compared with those who received placebo. In all trials, response to 2.5 mg was numerically greater than response to 1 mg and in the largest of the 3 trials, there was a statistically significant greater percentage of patients with headache response at 4 hours in the 2.5-mg group compared with the 1-mg group. The results are summarized in Table 2.
Table 2. Percentage of Adult Patients with Headache Response (Mild or No Headache) 4 Hours following Treatment
| AMERGE 1 mg (n=491) | AMERGE 2.5 mg (n=493) | Placebo (n=395) | |
|---|---|---|---|
| Trial 1 | 50%a | 60%a | 34% |
| Trial 2 | 52%a | 66%ab | 27% |
| Trial 3 | 54%a | 65%a | 32% |
a P<0.05 compared with placebo.
b P<0.05 compared with 1 mg.
The estimated probability of achieving an initial headache response in adults over the 4 hours following treatment in pooled Trials 1, 2, and 3 is depicted in Figure 1.
Figure 1. Estimated Probability of Achieving Initial Headache Response within 4 Hours in Pooled Trials 1, 2, and 3a:
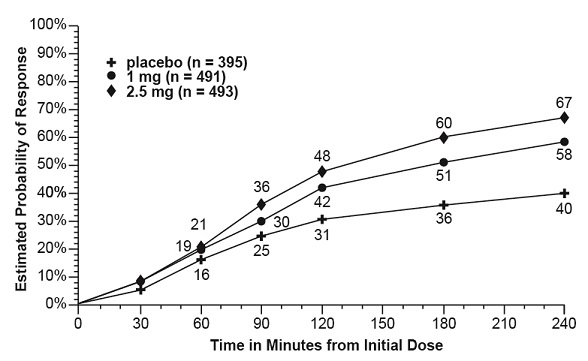
a The figure shows the probability over time of obtaining headache response (reduction in headache severity from moderate or severe pain to no or mild pain) following treatment with AMERGE. In this Kaplan-Meier plot, patients not achieving response within 240 minutes were censored at 240 minutes.
For patients with migraine-associated nausea, photophobia, and phonophobia at baseline, there was a lower incidence of these symptoms 4 hours following administration of 1-mg and 2.5-mg AMERGE compared with placebo.
Four to 24 hours following the initial dose of study treatment, patients were allowed to use additional treatment for pain relief in the form of a second dose of study treatment or other rescue medication. The estimated probability of patients taking a second dose or other rescue medication to treat migraine over the 24 hours following the initial dose of study treatment is summarized in Figure 2.
Figure 2. Estimated Probability of Patients Taking a Second Dose of AMERGE Tablets or Other Medication to Treat Migraine over the 24 Hours following the Initial Dose of Study Treatment in Pooled Trials 1, 2, and 3a:
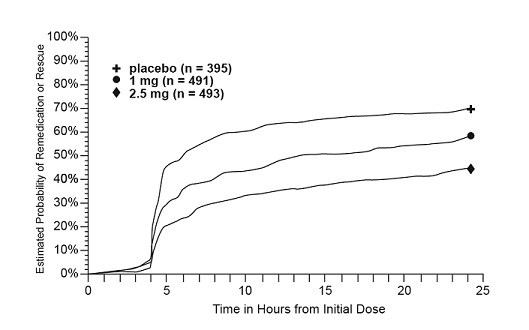
a Kaplan-Meier plot based on data obtained in the 3 controlled clinical trials (Trials 1, 2, and 3) providing evidence of efficacy with patients not using additional treatments censored at 24 hours. The plot also includes patients who had no response to the initial dose. Remedication was discouraged prior to 4 hours postdose.
There is no evidence that doses of 5 mg provided a greater effect than 2.5 mg. There was no evidence to suggest that treatment with AMERGE was associated with an increase in the severity or frequency of migraine attacks. The efficacy of AMERGE was unaffected by presence of aura; gender, age, or weight of the subject; oral contraceptive use; or concomitant use of common migraine prophylactic drugs (e.g., beta-blockers, calcium channel blockers, tricyclic antidepressants). There was insufficient data to assess the impact of race on efficacy.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.