AMVUTTRA Solution for injection Ref.[50230] Active ingredients: Vutrisiran
Source: European Medicines Agency (EU) Revision Year: 2025 Publisher: Alnylam Netherlands B.V., Antonio Vivaldistraat 150, 1083 HP Amsterdam, Netherlands
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Other Nervous System Drugs
ATC code: N07XX18
Mechanism of action
Amvuttra contains vutrisiran, a chemically stabilized double-stranded small interfering ribonucleic acid (siRNA) that specifically targets variant and/or wild-type transthyretin (TTR) messenger RNA (mRNA) and is covalently linked to a ligand containing three N-acetylgalactosamine (GalNAc) residues to enable delivery of the siRNA to hepatocytes.
Through a natural process called RNA interference (RNAi), vutrisiran causes the catalytic degradation of TTR mRNA in the liver, resulting in the reduction of serum levels of variant and wild-type amyloidogenic TTR protein thus reducing the deposition of TTR amyloid in tissues.
Pharmacodynamic effects
In HELIOS-A, mean serum TTR was rapidly reduced as early as Day 22, with mean near to steady state TTR reduction of 73% by Week 6. With repeat dosing of 25 mg once every 3 months, mean reductions of serum TTR after 9 and 18 months of treatment were 83% and 88%, respectively. Similar TTR reductions were observed regardless of genotype (V30M or non-V30M), prior TTR stabiliser use, weight, sex, age, or race.
In HELIOS-B, the mean serum TTR reduction profile was consistent with that observed in HELIOS-A, and similar across all subgroups studied (age, sex, race, body weight, anti-drug antibody [ADA] status, ATTR disease type [wild-type or hereditary], NYHA class, and baseline tafamidis use).
Serum TTR is a carrier of retinol binding protein 4, which is the principal carrier of vitamin A in the blood. In HELIOS-A, Amvuttra decreased serum vitamin A levels with mean steady state peak and trough reductions of 70% and 63%, respectively (see sections 4.4 and 4.5). In HELIOS-B, serum vitamin A reductions were consistent with those observed in HELIOS-A.
In HELIOS-B, NT-proBNP and Troponin I, cardiac biomarkers associated with heart failure, demonstrated relative stability in Amvuttra-treated patients for median change from baseline through Month 30 in the overall population (NT-proBNP: 9% increase; Troponin I: 10% decrease) while levels in placebo patients demonstrated worsening (NT-proBNP: 52% increase; Troponin I: 22% increase). Consistent trends were observed in the monotherapy population.
In HELIOS-B, centrally-assessed echocardiograms showed reduction relative to placebo favouring Amvuttra in LV wall thickness (LS mean difference: -0.4 mm [95% CI -0.8, -0.0]) and longitudinal strain (LS mean difference: -1.23% [95% CI -1.73, -0.73]) in the overall population. Results in the monotherapy population were consistent.
Clinical efficacy and safety
hATTR amyloidosis with polyneuropathy
The efficacy of Amvuttra was studied in a global, randomised, open-label clinical study (HELIOS-A) in adult patients with hATTR-PN. Patients were randomised 3:1 to receive 25 mg of Amvuttra (N=122) subcutaneously once every 3 months, or 0.3 mg/kg patisiran (N=42) intravenously once every 3 weeks. The treatment period of the study was conducted over 18 months with two analyses at Month 9 and at Month 18. Ninety-seven percent (97%) of Amvuttra-treated patients completed at least 18 months of the assigned treatments (vutrisiran or patisiran). Efficacy assessments were based on a comparison of the vutrisiran arm of the study with an external placebo group (placebo arm of the APOLLO Phase 3 study) comprised of a similar population of patients with hATTR-PN. Assessment of non-inferiority of serum TTR reduction was based on comparison of the vutrisiran arm to the within-study patisiran arm.
Of the patients who received Amvuttra, the median patient age at baseline was 60 years (range 34 to 80 years), 38% were ≥65 years old, and 65% of patients were male. Twenty-two (22) different TTR variants were represented: V30M (44%), T60A (13%), E89Q (8%), A97S (6%), S50R (4%), V122I (3%), L58H (3%), and Other (18%). Twenty percent (20%) of patients had the V30M genotype and early onset of symptoms (<50 years old). At baseline, 69% of patients had stage 1 disease (unimpaired ambulation; mild sensory, motor, and autonomic neuropathy in the lower limbs), and 31% had stage 2 disease (assistance with ambulation required; moderate impairment of the lower limbs, upper limbs, and trunk). There were no patients with stage 3 disease. Sixty-one percent (61%) of patients had prior treatment with TTR tetramer stabilisers. According to the New York Heart Association (NYHA) classification of heart failure, 9% of patients had class I and 35% had class II. Thirty-three percent (33%) of patients met pre-defined criteria for cardiac involvement (baseline LV wall thickness ≥13 mm with no history of hypertension or aortic valve disease).
The primary efficacy endpoint was the change from baseline to Month 18 in modified Neuropathy Impairment Score +7 (mNIS+7). This endpoint is a composite measure of motor, sensory, and autonomic neuropathy including assessments of motor strength, reflexes, quantitative sensory testing, nerve conduction studies, and postural blood pressure, with the score ranging from 0 to 304 points, where an increasing score indicates worsening impairment.
The change from baseline to Month 18 in Norfolk Quality of Life-Diabetic Neuropathy (QoL-DN) total score was assessed as a secondary endpoint. The Norfolk QoL-DN questionnaire (patient-reported) includes domains relating to small fibre, large fibre, and autonomic nerve function, symptoms of polyneuropathy, and activities of daily living, with the total score ranging from -4 to 136, where increasing score indicates worsening quality of life.
Other secondary endpoints included gait speed (10-meter walk test), nutritional status (mBMI), and patient-reported ability to perform activities of daily living and social participation (Rasch-Built Overall Disability Scale [R-ODS]).
Treatment with Amvuttra in the HELIOS-A study demonstrated statistically significant improvements in all endpoints (Table 2 and Figure 1) measured from baseline to Month 9 and 18, compared to the external placebo group of the APOLLO study (all p<0.0001).
The time-averaged trough TTR percent reduction through Month 18 was 84.7% for vutrisiran and 80.6% for patisiran. The percent reduction in serum TTR levels in the vutrisiran arm was non-inferior (according to predefined criteria) to the within-study patisiran arm through Month 18 with a median difference of 5.3% (95% CI 1.2%, 9.3%).
Table 2. Summary of clinical efficacy results from the HELIOS-A study:
| Endpointa | Baseline, Mean (SD) | Change from Baseline, LS Mean (SEM) | Amvuttra-Placebob Treatment Difference, LS Mean (95% CI) | p-value | ||
|---|---|---|---|---|---|---|
| Amvuttra N=122 | Placebob N=77 | Amvuttra | Placebob | |||
| Month 9 | ||||||
| mNIS+7c | 60.6 (36.0) | 74.6 (37.0) | -2.2 (1.4) | 14.8 (2.0) | -17.0 (-21.8, -12.2) | p<0.0001 |
| Norfolk QoL-DNc | 47.1 (26.3) | 55.5 (24.3) | -3.3 (1.7) | 12.9 (2.2) | -16.2 (-21.7, -10.8) | p<0.0001 |
| 10-meter walk test (m/sec)d | 1.01 (0.39) | 0.79 (0.32) | 0 (0.02) | -0.13 (0.03) | 0.13 (0.07, 0.19) | p<0.0001 |
| Month 18 | ||||||
| mNIS+7c | 60.6 (36.0) | 74.6 (37.0) | -0.5 (1.6) | 28.1 (2.3) | -28.5 (-34.0, -23.1) | p<0.0001 |
| Norfolk QoL-DNc | 47.1 (26.3) | 55.5 (24.3) | -1.2 (1.8) | 19.8 (2.6) | -21.0 (-27.1, -14.9) | p<0.0001 |
| 10-meter walk test (m/sec)d | 1.01 (0.39) | 0.79 (0.32) | -0.02 (0.03) | -0.26 (0.04) | 0.24 (0.15, 0.33) | p<0.0001 |
| mBMIe | 1057.5 (233.8) | 989.9 (214.2) | 25.0 (9.5) | -115.7 (13.4) | 140.7 (108.4, 172.9) | p<0.0001 |
| R-ODSf | 34.1 (11.0) | 29.8 (10.8) | -1.5 (0.6) | -9.9 (0.8) | 8.4 (6.5, 10.4) | p<0.0001 |
Abbreviations: CI = confidence interval; LS mean = least squares mean; mBMI = modified body mass index; mNIS = modified Neuropathy Impairment Score; QoL-DN = Quality of Life - Diabetic Neuropathy; SD = standard deviation; SEM = standard error of the mean
a All Month 9 endpoints analysed using the analysis of covariance (ANCOVA) with multiple imputation (MI) method and all Month 18 analysed using the mixed-effects model for repeated measures (MMRM)
b External placebo group from APOLLO randomised controlled study
c A lower number indicates less impairment/fewer symptoms
d A higher number indicates less disability/less impairment
e mBMI: body mass index (BMI; kg/m²) multiplied by serum albumin (g/L); a higher number indicates better nutritional status.
f A higher number indicates less disability/less impairment.
Figure 1. Change from Baseline in mNIS+7 (Month 9 and Month 18):
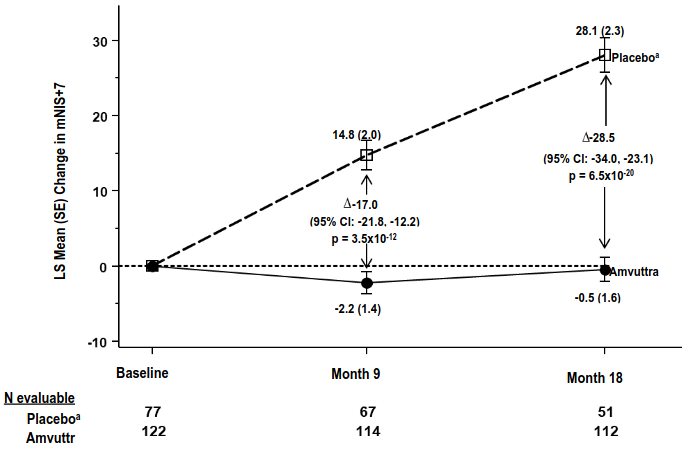
A decrease in mNIS+7 indicates improvement
∆ indicates between-group treatment difference, shown as the LS mean difference (95% CI) for AMVUTTRA–external placebo
All Month 9 endpoints analysed using the analysis of covariance (ANCOVA) with multiple imputation (MI) method and all Month 18 analysed using the mixed-effects model for repeated measures (MMRM)
a External placebo group from APOLLO randomised controlled study
Patients receiving Amvuttra experienced similar benefit relative to placebo in mNIS+7 and Norfolk QoL-DN total score at Month 9 and Month 18 across all subgroups including age, sex, race, region, NIS score, V30M genotype status, prior TTR stabiliser use, disease stage, and patients with or without pre-defined criteria for cardiac involvement.
The N-terminal prohormone-B-type natriuretic peptide (NT-proBNP) is a prognostic biomarker of cardiac dysfunction. NT-proBNP- baseline values (geometric mean) were 273 ng/L and 531 ng/L in Amvuttra-treated and placebo-treated patients, respectively. At Month 18, the geometric mean NT-proBNP levels decreased by 6% in Amvuttra patients, while there was a 96% increase in placebo patients.
Centrally-assessed echocardiograms showed changes in LV wall thickness (LS mean difference: -0.18 mm [95% CI -0.74, 0.38]) and longitudinal strain (LS mean difference: -0.4% [95% CI -1.2, 0.4]) with Amvuttra treatment relative to placebo.
wtATTR or hATTR amyloidosis with cardiomyopathy
The efficacy of Amvuttra was demonstrated in a global, randomised, double-blind, placebo-controlled clinical study (HELIOS-B) in adult patients with ATTR-CM. Patients were randomized 1:1 to receive 25 mg of Amvuttra subcutaneously once every 3 months, or matching placebo. At baseline, 40% of patients were receiving treatment with tafamidis. Treatment assignment was stratified by baseline tafamidis use, ATTR disease type (wtATTR or hATTR amyloidosis), and by baseline severity of disease and age (NYHA Class I or II and age <75 years versus all other).
Of the patients who received Amvuttra, at baseline, the median patient age was 77 years (range 45 to 85 years) and 92% were male. Eighty five percent (85%) of patients were Caucasian, 7% were Black or African American, 6% were Asian. Eighty nine percent (89%) of patients had wtATTR amyloidosis and 11% had hATTR amyloidosis. According to the NYHA classification of heart failure (HF), 15% of patients had Class I, 77% had Class II, and 8% had Class III and were NAC ATTR disease stage 1 or 2. Patient demographics and baseline disease characteristics were similar between the treatment groups.
The primary efficacy endpoint was the composite outcome of all-cause mortality and recurrent CV events (CV hospitalisations and urgent heart failure [UHF] visits) during the double-blind treatment period of up to 36 months, evaluated in the overall population and in the monotherapy population (defined as patients not receiving tafamidis at study baseline).
Amvuttra led to significant reductions in the risk of all-cause mortality and recurrent CV events compared to placebo in the overall and monotherapy populations of 28.2% and 32.8%, respectively (Table 3). Approximately 77% of all deaths in HELIOS-B were CV-related. The rate of both CV deaths and non-CV deaths was lower in Amvuttra-treated patients compared to placebo. Of the total number of CV events, 87.9% were CV hospitalisations, and 12.1% were UHF visits. A Kaplan-Meier curve illustrating time to first CV event or all-cause mortality is presented in Figure 2.
Both components of the primary composite endpoint individually contributed to the treatment effect in the overall population and monotherapy population (Table 3).
In the secondary endpoint analysis of all-cause mortality including data up to Month 42, incorporating the double-blind period and up to an additional 6 months of survival data for all patients, Amvuttra led to a 35.5% reduction in the risk of death relative to placebo in the overall population (hazard ratio: 0.645; 95% CI: 0.463, 0.898; p=0.0098), and to a 34.5% reduction in the monotherapy population (hazard ratio: 0.655; 95% CI: 0.440, 0.973; p=0.0454).
Table 3. Primary composite endpoint and its individual components in HELIOS-B:
| Endpoint | Overall population | Monotherapy population | |||
|---|---|---|---|---|---|
| Amvuttra (N=326) | Placebo (N=328) | Amvuttra (N=196) | Placebo (N=199) | ||
| Primary composite endpointa | Hazard Ratio (95% CI)b p-valueb | 0.718 (0.555, 0.929) 0.0118 | 0.672 (0.487, 0.929) 0.0162 | ||
| Components of the Primary Composite Endpoint | |||||
| All-cause mortality | Hazard Ratio (95% CI)c | 0.694 (0.490, 0.982) | 0.705 (0.467, 1.064) | ||
| CV hospitalisations and UHF visits | Relative Rate Ratio (95% CI)d | 0.733 (0.610, 0.882) | 0.676 (0.533, 0.857) | ||
Abbreviations: CI=confidence interval; CV=cardiovascular; UHF=urgent heart failure
Heart transplantation and left ventricular assist device placement are treated as death. Deaths after study discontinuation are included in the all-cause mortality component analysis.
a Primary composite endpoint defined as: composite outcome of all-cause mortality and recurrent CV events. Primary analysis included at least 33 months (and up to 36 months) follow-up on all patients.
b Hazard Ratio (95% CI) and p-value are based on a modified Andersen-Gill model.
c Hazard Ratio (95% CI) is based on a Cox proportional hazard model.
d Relative rate ratio (95% CI) is based on a Poisson regression model.
Figure 2. Time to First CV Event or All-Cause Mortality (Overall population):

Abbreviation: CI = confidence interval; CV = cardiovascular; HR = hazard ratio.
Heart transplantation and left ventricular assist device placement are treated as death. Kaplan-Meier curves are adjusted for baseline disease characteristics using the inverse probability of treatment weighting method. HR and 95% CI are based on a Cox proportional hazard model, and p-value is based on log-rank test.
Results from the subgroup analysis for the primary composite endpoint favoured Amvuttra across all prespecified subgroups in the overall population and the monotherapy population. In the subgroup of patients on background tafamidis, Amvuttra led to a 21.5% numerical reduction in the risk of all-cause mortality and recurrent CV events relative to placebo (hazard ratio: 0.785; 95% CI: 0.511, 1.207) (Figure 3).
Figure 3. Subgroup Analyses of the Primary Composite Endpoint (Overall Population):

Abbreviations: ATTR = transthyretin amyloidosis; CI = confidence interval; hATTR = hereditary transthyretin amyloidosis; HR = hazard ratio; NT-proBNP = N-terminal prohormone of B-type natriuretic peptide; NYHA = New York Heart Association; wtATTR = wild-type transthyretin amyloidosis.
HR and 95% CI are based on modified Andersen-Gill model analyses.
The treatment effects of Amvuttra on functional capacity, patient-reported health status and quality of life, and heart failure symptom severity were assessed by the change from baseline to Month 30 in 6-Minute Walk Test (6-MWT), the Kansas City Cardiomyopathy Questionnaire-Overall Summary (KCCQ-OS) score and NYHA class, respectively. The KCCQ-OS is composed of four domains including Total Symptoms (Symptom Frequency and Symptom Burden), Physical Limitation, Quality of Life, and Social Limitation. The Overall Summary score and domain scores range from 0 to 100, with higher scores representing better health status.
A statistically significant treatment effect favouring Amvuttra was observed for 6-MWT distance, KCCQ-OS score, and stable or improved NYHA class, in both the overall population and monotherapy population (Table 4), with consistent results across all subgroups studied. The treatment effect on KCCQ-OS score was consistent across all four domain scores.
Table 4. Change from Baseline in 6-MWT distance, KCCQ-OS score and NYHA class at Month 30:
| Overall population | Monotherapy population | |||
|---|---|---|---|---|
| Amvuttra (N=326) | Placebo (N=328) | Amvuttra (N=196) | Placebo (N=199) | |
| 6-MWT (metres) | ||||
| Baseline Mean (SD) | 372 (104) | 377 (96) | 363 (103) | 373 (98) |
| Change from baseline to Month 30, LS Mean (SE)a | -45 (5) | -72 (5) | -60 (7) | -92 (6) |
| Treatment Difference from Placebo, LS Mean (95% CI) p-valuea,b | 26 (13, 40) <0.0001 | 32 (14, 50) 0.0005 | ||
| KCCQ-OS (points) | ||||
| Baseline Mean (SD) | 73 (19) | 72 (20) | 70 (20) | 70 (21) |
| Change from baseline to Month 30, LS Mean (SE)a | -10 (1) | -15 (1) | -11 (2) | -19 (2) |
| Treatment Difference from Placebo, LS Mean (95% CI) p-valuea,b | 6 (2, 9) 0.0008 | 9 (4, 13) 0.0003 | ||
| NYHA Class | ||||
| % of patients with stable or improved NYHA class at Month 30 | 68 | 61 | 66 | 56 |
| Difference from Placebo, (%) (95% CI)c p-valuec | 9 (1, 16) 0.0217 | 13 (3, 22) 0.0121 | ||
Abbreviations: 6-MWT = 6-minute walk test; KCCQ-OS = Kansas City Cardiomyopathy Questionnaire, LS = least squares; CI = confidence interval; SD = Standard deviation; SE = Standard Error; NYHA = New York Heart Association
a For assessment missing because of death (including heart transplantation and left ventricular assist device placement), and inability to walk as the result of ATTR disease progression (applicable to 6-MWT only), data were imputed from resampling of the worst 10% of observed changes.
b Estimated from the MMRM (mixed-effect model repeated measures) model.
c Based on Cochran-Mantel-Haenszel method.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with vutrisiran in all subsets of the paediatric population in hATTR amyloidosis (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
The pharmacokinetic properties of Amvuttra were characterised by measuring the plasma and urine concentrations of vutrisiran.
Absorption
Following subcutaneous administration, vutrisiran is rapidly absorbed with a time to maximum plasma concentration (tmax) of 3.0 (range: 2.0 to 6.5) hours. At the recommended dosing regimen of 25 mg once every 3 months subcutaneously, the mean (% coefficient of variation [%CV]) steady state peak concentrations (Cmax), and area under the concentration time curve from 0 to 24 hours (AUC0-24) were 0.12 μg/mL (64.3%), and 0.80 μg·h/mL (35.0%), respectively. There was no accumulation of vutrisiran in plasma after repeated quarterly dosing.
Distribution
Vutrisiran is greater than 80% bound to plasma proteins over the concentration range observed in humans at the dose of 25 mg once every 3 months subcutaneously. Vutrisiran plasma protein binding was concentration-dependent and decreased with increasing vutrisiran concentrations (from 78% at 0.5 μg/mL to 19% at 50 μg/mL). The population estimate for the apparent central compartment volume of distribution (Vd/F) of vutrisiran in humans was 10.2 L (% Relative standard error [RSE]=5.71%). Vutrisiran distributes primarily to the liver after subcutaneous dosing.
Biotransformation
Vutrisiran is metabolised by endo- and exo-nucleases to short nucleotide fragments of varying sizes within the liver. There were no major circulating metabolites in humans. In vitro studies indicate that vutrisiran does not undergo metabolism by CYP450 enzymes.
Elimination
Following a 25 mg single subcutaneous dose, the median apparent plasma clearance was 21.4 (range: 19.8, 30.0) L/h. The median terminal elimination half-life (t1/2) of vutrisiran was 5.23 (range: 2.24, 6.36) hours. After a single subcutaneous dose of 5 to 300 mg, the mean fraction of unchanged active substance eliminated in urine ranged from 15.4 to 25.4% and the mean renal clearance ranged from 4.45 to 5.74 L/h for vutrisiran.
Linearity/non-linearity
Following single subcutaneous doses over the 5 to 300 mg dose range, vutrisiran Cmax was shown to be dose proportional while area under the concentration-time curve from the time of dosing extrapolated to infinity (AUCinf) and area under the concentration-time curve from the time of dosing to the last measurable concentration (AUClast) were slightly more than dose proportional.
Pharmacokinetic/pharmacodynamic relationship(s)
Population pharmacokinetic/pharmacodynamic analyses in healthy subjects and patients with hATTR amyloidosis (n=202) showed a dose-dependent relationship between predicted vutrisiran liver concentrations and reductions in serum TTR. The model-predicted median steady state peak, trough, and average TTR reductions were 88%, 86%, and 87%, respectively, confirming minimal peak-to-trough variability across the 3-month dosing interval. Covariate analysis indicated similar TTR reduction in patients with mild-to-moderate renal impairment or mild hepatic impairment, as well as by sex, race, prior use of TTR stabilisers, genotype (V30M or non-V30M), age and weight.
Special populations
Gender and race
Clinical studies did not identify significant differences in steady state pharmacokinetic parameters or TTR reduction according to gender or race.
Elderly patients
In the HELIOS-A study, 46 (38%) patients treated with vutrisiran were ≥65 years old and of these 7 (5.7%) patients were ≥75 years old. In the HELIOS-B study, 299 (91.7%) patients treated with vutrisiran were ≥65 years old, with a median age of 77.0 years, and of these 203 (62.3%) were ≥75 years old. There were no significant differences in steady state pharmacokinetic parameters or TTR reduction.
Hepatic impairment
Clinical studies indicated no impact of mild (total bilirubin ≤1 x ULN and AST >1 x ULN, or total bilirubin >1.0 to 1.5 x ULN and any AST) or moderate (total bilirubin >1.5 to 3 × ULN and any AST) hepatic impairment on vutrisiran exposure or TTR reduction compared to patients with normal hepatic function. Vutrisiran has not been studied in patients with severe hepatic impairment.
Renal impairment
Clinical studies indicated no impact of mild or moderate renal impairment (eGFR ≥30 to <90 mL/min/1.73 m²) on vutrisiran exposure or TTR reduction compared to subjects with normal renal function. Vutrisiran has not been studied in patients with severe renal impairment or end-stage renal disease.
5.3. Preclinical safety data
General toxicology
Repeated once-monthly subcutaneous administration of vutrisiran at ≥ 30 mg/kg in monkeys produced the expected sustained reductions of circulating TTR (up to 99%) and vitamin A (up to 89%) without any apparent toxicological findings.
Following once monthly repeated dosing for up to 6 months in rats and 9 months in monkeys, the mild and consistent non-adverse histological changes in liver (hepatocytes, Kupffer cells), kidneys (renal tubules), lymph nodes and injection sites (macrophages) reflected the principal distribution and accumulation of vutrisiran. However, no toxicities were identified at up to more than 1 000- and 3 000-fold higher plasma AUC, when normalised to quarterly dosing and compared to the anticipated exposure at the maximum recommended human dose [MRHD].
Genotoxicity/Carcinogenicity
Vutrisiran did not exert any genotoxic potential in vitro and in vivo. Vutrisiran was not carcinogenic in rats and in male mice. In female mice dosed once monthly with vutrisiran at 3, 9, or 18 mg/kg, a statistically significant dose-dependent trend for combined hepatocellular adenomas and carcinomas was observed with unknown relevance for humans. The carcinogenic potential of vutrisiran is considered low if all toxicity data are taken into account.
Reproductive toxicity
Vutrisiran is not pharmacologically active in rats and rabbits, which limits the predictivity of these investigations. Nevertheless, a single dose of a rat-specific orthologue of vutrisiran did not impact on fertility and early embryonic development in a combined study in rats.
Weekly subcutaneous administrations of vutrisiran did not affect fertility and early embryonic development at more than 300-times the normalised MRHD In an embryo-foetal study with daily subcutaneous vutrisiran administration in pregnant rats, adverse effects on maternal body weight, food consumption, increased premature delivery and post-implantation loss were observed with a maternal NOAEL of 10 mg/kg/day that was more than 300-times the normalised MRHD of 0.005 mg/kg/day. Based on an adverse reduction in foetal body weights and increased skeletal variations at ≥10 mg/kg/day, the foetal NOAEL of vutrisiran was 3 mg/kg/day which is 97-times the normalised MRHD.
In an embryo-foetal development study in pregnant rabbits, no adverse effects on embryo-foetal development were observed at ≤30 mg/kg/day vutrisiran, which is more than 1 900-times the normalised MRHD.
In a prenatal-postnatal development study, subcutaneous vutrisiran administration on every 6 th day had no effect on growth and development of the offspring with a NOAEL of 20 mg/kg, which was more than 90-times the normalised MRHD.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.