AQNEURSA Granules for oral suspension Ref.[116287] Active ingredients: Levacetylleucine
Source: European Medicines Agency (EU) Revision Year: 2026 Publisher: IntraBio Ireland Ltd, 10 Earlsfort Terrace, Dublin 2, Ireland
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Other nervous system drugs
ATC code: N07XX27
Pharmacodynamic effects
Aqneursa contains levacetylleucine that targets underlying processes of neurological dysfunction. Nonclinical studies demonstrated that levacetylleucine corrects energy metabolism, including improving adenosine triphosphate production.
Clinical efficacy and safety
The efficacy and safety of levacetylleucine for treatment of NPC was studied in a randomised, double- blind, placebo-controlled, 2-period crossover study that evaluated the efficacy of levacetylleucine in NPC patients. To be eligible for the study, patients had to be aged 4 years or older with a confirmed diagnosis of NPC, a baseline SARA score of 7 to 34 points, and not receiving any other investigational therapies; patients were permitted to be receiving treatment with miglustat.
Patients were randomised in a 1:1 ratio to receive either levacetylleucine or placebo for 12 weeks in Period I. In Period II, patients switched to the opposite (either levacetylleucine or placebo) for 12 weeks. Patients aged ≥ 13 years received 4 g/day (as 2 g morning dose, 1 g afternoon dose and 1 g evening dose). The levacetylleucine dose in children under 13 years was based on patient's body weight (see section 4.2).
A total of 60 patients were randomised and treated, and 59 (98%) completed both treatment periods with levacetylleucine and placebo.
Of the 60 randomised patients (37 adults and 23 paediatric patients), 27 were female and 33 were male. The median age at treatment initiation was 25 years (range: 5 to 67 years). 90% of the patients were White, 3% Asian and 7% Other. In total, 51 (85%) patients received miglustat treatment prior to randomisation and during the study.
The primary endpoint measure was the measurement of neurological signs, symptoms and functioning as measured by the Scale for the Assessment and Rating of Ataxia (SARA).
The SARA assessment was carried out at baseline and then assessed at the end of Period I (Week 12) and the end of Period II (Week 24). Treatment with levacetylleucine demonstrated a statistically significant difference in favour of levacetylleucine as compared to placebo on SARA (Table 3).
Table 3. Summary of scale for the assessment and rating of ataxia (SARA) efficacy results*:
| Effect/Variable | Mean difference (SD) | Estimate (SE) | 95% CI | p-value** |
| Baseline value | -- | 0.95 (0.04) | (0.86, 1.04) | <0.001 |
| Treatment effect (levacetylleucine versus placebo) | -- | -1.28 (0.31) | (-1.91, -0.65) | <0.001 |
| Levacetylleucine total change versus baseline | -1.97 (2.43) | -- | -- | -- |
| Placebo total change versus baseline | -0.60 (2.39) | -- | -- | -- |
CI = confidence interval; SD = standard deviation; SE = standard error.
* The change from baseline was assessed at the end of Period 1 (Week 12) and the end of Period 2 (Week 24).
** Two-sided p-value
The 30 patients receiving placebo in Period I had no meaningful change in the mean SARA score of -0.60 compared to those on levacetylleucine, who had a substantial change of -1.93. The remaining 30 patients who received levacetylleucine in Period I followed by placebo in Period II experienced significant worsening of symptoms on placebo, which effectively served as a washout from levacetylleucine (difference in mean SARA score between the end of Period I [Week 12] and the end of Period II [Week 24] of +1.55), reflecting a deterioration in neurological signs and symptoms when treatment with levacetylleucine was stopped (Figure 1).
In total, 9 patients (15%) were not on miglustat prior to randomisation and during the study. In these patients, there was also a change in the mean SARA score on levacetylleucine of -2.06.
Figure 1. Individual patient SARA total scores at baseline, end of Period I, and End of Period II:
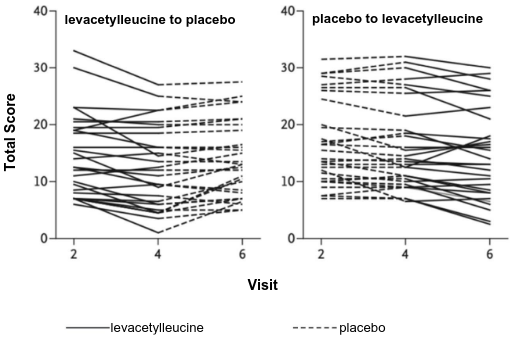
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with levacetylleucine in one or more subsets of the paediatric population in the treatment of Niemann-Pick disease, type C (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Absorption
After oral administration, levacetylleucine is rapidly absorbed. Median time to maximum concentration (Cmax), tmax, is 1 hour (ranging from 0.5 to 2.5 hours). Mean, dose-normalised (per gram of levacetylleucine) Cmax and area under the curve from time 0 to 24 hours (AUC0-24hrs) were 4 mcg/mL/g and 9 h*mcg/mL/g. The absolute oral bioavailability is unknown.
No studies on food effect on absorption have been conducted.
Distribution
The mean (standard deviation [SD]) volume of distribution at steady state (Vss) was 253 (125) L. Levacetylleucine is taken up by ubiquitously expressed monocarboxylate transporters, thereby delivering levacetylleucine to all tissues including the central nervous system.
Elimination
The mean (SD) clearance is 139 (59) L/h. The estimated half-life is around 1 hour. No or only minor accumulation was observed after repeat administration.
Characteristics in specific groups of subjects or patients
There were no clinically significant differences in the PK of levacetylleucine based on age (range: 6-67 years), sex, race/ethnicity or body weight (range: 20.5-98.4 kg).
Renal or hepatic impairment
The effect of renal or hepatic impairment on the PK of levacetylleucine has not been studied.
Interaction with other medicinal products
In vitro studies showed that levacetylleucine, at therapeutic concentrations, does not significantly inhibit the enzyme activity of cytochrome P450 (CYP450) isoforms. Levacetylleucine does not have induction potential towards CYP450 enzymes.
In vitro, levacetylleucine inhibited the efflux transporters P-glycoprotein (P-gp), breast cancer resistance protein (BCRP) or bile salt export pump (BSEP) (see section 4.5).
Levacetylleucine is a substrate and an in vitro inhibitor of organic anion transporter (OAT)1 and OAT3. The likelihood of clinically meaningful drug interactions is considered low.
5.3. Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity and genotoxicity.
No effect on fertility was seen in rats at doses up to 1 000 mg/kg/day (1.5- to 2.3-fold human exposure). In embryofoetal development studies, levacetylleucine did not induce adverse developmental effects at doses up to 1 000 mg/kg/day in rats (2.0-fold human exposure). In rabbits, external and skeletal malformations were observed at 1 250 mg/kg/day (7.1-fold of human exposure) with a no observed adverse effect level of 675 mg/kg/day (4.9-fold human exposure). No adverse effects were observed in a pre- and postnatal development study in rats at doses up to 1 000 mg/kg/day.
Carcinogenesis
No carcinogenicity studies have been conducted. The carcinogenic risk is unknown.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.