ASPAVELI Solution for infusion Ref.[49950] Active ingredients: Pegcetacoplan
Source: European Medicines Agency (EU) Revision Year: 2026 Publisher: Swedish Orphan Biovitrum AB (publ), SE-112 76 Stockholm, Sweden
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Immunosuppressants, Complement inhibitor
ATC code: L04AJ03
Mechanism of action
Pegcetacoplan is a symmetrical molecule comprised of two identical pentadecapeptides covalently bound to the ends of a linear 40-kDa PEG molecule. The peptide moieties bind to complement C3 and C3b and exert a broad inhibition of the complement cascade. The 40-kDa PEG moiety imparts improved solubility and longer residence time in the body after administration of the medicinal product.
Pegcetacoplan binds to complement protein C3 and its activation fragment C3b with high affinity, thereby regulating the cleavage of C3 and the generation of downstream effectors of complement activation. In PNH, extravascular haemolysis (EVH) is facilitated by C3b opsonisation while intravascular haemolysis (IVH) is mediated by the downstream membrane attack complex (MAC). Pegcetacoplan exerts broad regulation of the complement cascade by acting proximal to both C3b and MAC formation, thereby controlling the mechanisms that lead to EVH and IVH.
In C3G and primary IC-MPGN, there is excessive deposition of C3 breakdown products in the glomeruli of the kidney leading to renal parenchymal damage and impairment of kidney function. Pegcetacoplan targets upstream effectors of complement activation (C3 and C3b), thereby inhibiting activation initiated by all (alternative, classical and lectin) complement pathways. By inhibiting C3, pegcetacoplan directly addresses the inappropriate C3 activation and modifies the underlying disease by reducing the excessive deposition of C3 breakdown products in the glomeruli of the kidney. By targeting C3b, pegcetacoplan also inhibits the activity of the alternative pathway (AP) C3 convertase through an additional mechanism of action in the complement cascade. This further prevents deposition of C3 breakdown products in the glomeruli.
Pharmacodynamic effects
PNH
In Study APL2-302, the mean serum C3 concentration increased from 0.94 g/L at baseline to 3.83 g/L at Week 16 in the pegcetacoplan group and sustained through Week 48. In Study APL2-308, the mean serum C3 concentration increased from 0.95 g/L at baseline to 3.56 g/L at Week 26.
In Study APL2-302, the mean percentage of PNH Type II + III RBCs increased from 66.80% at baseline, to 93.85% at Week 16 and sustained through Week 48. In Study APL2-308, the mean percentage of PNH Type II + III RBCs increased from 42.4% at baseline to 90.0% at Week 26.
In Study APL2-302, the mean percentage of PNH Type II + III RBCs with C3 deposition was decreased from 17.73% at baseline to 0.20% at Week 16 and sustained through Week 48. In Study APL2-308, the mean percentage of PNH Type II + III RBCs with C3 deposition decreased from 2.85% at baseline to 0.09% at Week 26.
C3G and primary IC-MPGN
In Study APL2-C3G-310, the mean serum C3 concentration increased from 0.62 g/L at baseline to 3.71 g/L at Week 26 in the pegcetacoplan group and the effect was sustained up to Week 52. In the placebo group, C3 concentrations remained stable up to Week 26 (0.57 g/L at baseline; 0.58 g/L at Week 26) and increased upon switch to pegcetacoplan to 3.59 g/L at Week 52.
Mean serum sC5b-9 concentration decreased from 902.5 ng/mL at baseline to 290.2 ng/mL at Week 26 in the pegcetacoplan group and the effect was sustained up to Week 52. In the placebo group, sC5b-9 concentrations remained stable (768.3 ng/mL at baseline; 759.9 ng/mL at Week 26) and decreased upon switch to pegcetacoplan to 272.9 ng/mL at Week 52.
Clearance of glomerular C3 deposits at 6 months was observed based on a greater proportion of patients achieving a staining score of zero in the pegcetacoplan (71.4%) as compared to the placebo (8.8%) group.
Clinical efficacy and safety
PNH
The efficacy and safety of pegcetacoplan in patients with PNH was assessed in two open-label, randomised-controlled phase 3 studies: in complement inhibitor-experienced patients in Study APL2-302 and in complement inhibitor-naïve patients in Study APL2-308. In both studies the dose of pegcetacoplan was 1 080 mg twice weekly. If required, the dose could be adjusted to 1 080 mg every 3 days.
Study in complement inhibitor-experienced adult patients (APL2-302)
Study APL2-302 was an open-label, randomised study with an active comparator-controlled period of 16 weeks followed by a 32-week open label period (OLP). This study enrolled patients with PNH who had been treated with a stable dose of eculizumab for at least the previous 3 months and with haemoglobin levels <10.5 g/dL. Eligible patients entered a 4-week run-in period during which they received pegcetacoplan 1 080 mg subcutaneously twice weekly in addition to their current dose of eculizumab. Patients were then randomised in a 1:1 ratio to receive either 1 080 mg of pegcetacoplan twice weekly or their current dose of eculizumab through the duration of the 16-week randomised controlled period (RCP). Randomisation was stratified based on the number of packed red blood cell (PRBC) transfusions within the 12 months prior to Day -28 (<4; ≥4) and platelet count at screening (<100 000/mm³; ≥100 000/mm³). Patients who completed the RCP entered the OLP during which all patients received pegcetacoplan for up to 32 weeks (patients who received eculizumab during the RCP entered a 4-week run-in period before switching to pegcetacoplan monotherapy).
The primary and secondary efficacy endpoints were assessed at Week 16. The primary efficacy endpoint was change from baseline to Week 16 (during RCP) in Hb level. Baseline was defined as the average of measurements prior to the first dose of pegcetacoplan (at the beginning of the run-in period). Key secondary efficacy endpoints were transfusion avoidance, defined as the proportion of patients who did not require a transfusion during the RCP, and change from baseline to Week 16 in absolute reticulocyte count (ARC), LDH level, and functional assessment of chronic illness therapy (FACIT)-Fatigue scale score.
A total of 80 patients entered the run-in period. At the end of the run-in period, all 80 were randomised, 41 to pegcetacoplan and 39 to eculizumab. Demographics and baseline disease characteristics were generally well balanced between treatment groups (see Table 2). A total of 38 patients in the group treated with pegcetacoplan and 39 patients in the eculizumab group completed the 16-week RCP and continued into the 32-week open-label period. In total, 12 of 80 (15%) patients receiving pegcetacoplan discontinued due to adverse events. Per protocol 15 patients had their dose adjusted to 1 080 mg every 3 days. Twelve patients were evaluated for benefit and 8 of the 12 patients demonstrated benefit from the dose adjustment.
Table 2. Patient baseline demographics and characteristics in Study APL2-302:
| Parameter | Statistics | Pegcetacoplan (N=41) | Eculizumab (N=39) |
|---|---|---|---|
| Age (years) 18-64 years ≥65 years | Mean (SD) n (%) n (%) | 50.2 (16.3) 31 (75.6) 10 (24.4) | 47.3 (15.8) 32 (82.1) 7 (17.9) |
| Dose level of eculizumab at baseline Every 2 weeks IV 900 mg Every 11 days IV 900 mg Every 2 weeks IV 1 200 mg Every 2 weeks IV 1 500 mg | n (%) n (%) n (%) n (%) | 26 (63.4) 1 (2.4) 12 (29.3) 2 (4.9) | 29 (74.4) 1 (2.6) 9 (23.1) 0 |
| Female | n (%) | 27 (65.9) | 22 (56.4) |
| Time since diagnosis of PNH (years) to Day -28 | Mean (SD) | 8.7 (7.4) | 11.4 (9.7) |
| Haemoglobin level (g/dL) | Mean (SD) | 8.7 (1.1) | 8.7 (0.9) |
| Reticulocyte count (109/L) | Mean (SD) | 218 (75.0) | 216 (69.1) |
| LDH level (U/L) | Mean (SD) | 257.5 (97.6) | 308.6 (284.8) |
| Total FACIT-Fatigue* | Mean (SD) | 32.2 (11.4) | 31.6 (12.5) |
| Number of transfusions in last 12 months prior to Day -28 | Mean (SD) | 6.1 (7.3) | 6.9 (7.7) |
| <4 | n (%) | 20 (48.8) | 16 (41.0) |
| ≥4 | n (%) | 21 (51.2) | 23 (59.0) |
| Platelet count at screening (109/L) | Mean (SD) | 167 (98.3) | 147 (68.8) |
| Platelet count at screening <100 000/mm³ | n (%) | 12 (29.3) | 9 (23.1) |
| Platelet count at screening ≥100 000/mm³ | n (%) | 29 (70.7) | 30 (76.9) |
| History of aplastic anaemia | n (%) | 11 (26.8) | 9 (23.1) |
| History of myelodysplastic syndrome | n (%) | 1 (2.4) | 2 (5.1) |
* FACIT-Fatigue is measured on a scale of 0-52, with higher values indicating less fatigue.
Pegcetacoplan was superior to eculizumab for the primary endpoint of the haemoglobin change from baseline (P<0.0001).
Figure 1. Adjusted mean change in haemoglobin (g/dL) from baseline to Week 16 in APL2-302:
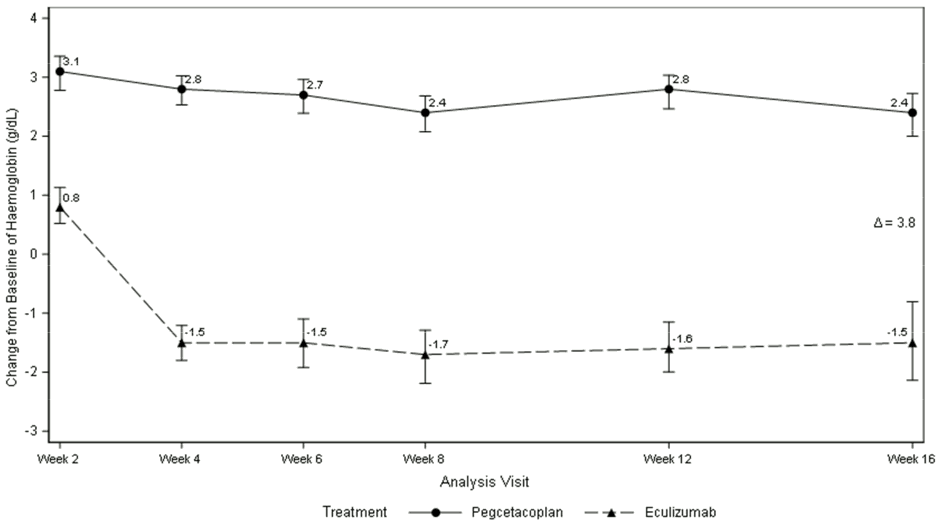
Non-inferiority was demonstrated in key secondary endpoints of transfusion avoidance and change from baseline in ARC.
Non-inferiority was not met in change from baseline in LDH.
Due to hierarchical testing, statistical testing for change from baseline for FACIT-Fatigue score was not formally tested.
The adjusted means, treatment difference, confidence intervals, and statistical analyses performed for the key secondary endpoints are shown in Figure 2.
Figure 2. Key secondary endpoints analysis in APL2-302:
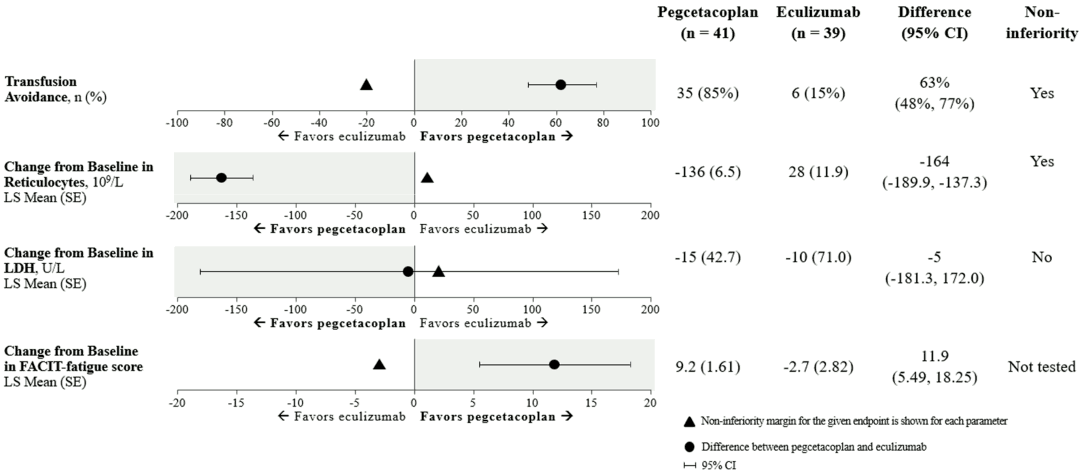
Results were consistent across all supportive analyses of the primary and key secondary endpoints, including all observed data with post transfusion data included.
Hb normalisation was achieved in 34% of patients in the pegcetacoplan group versus 0% in the eculizumab group at Week 16. LDH normalisation was achieved in 71% of patients in the group treated with pegcetacoplan versus 15% in the eculizumab group.
A total of 77 patients entered the 32-week OLP, during which all patients received pegcetacoplan, resulting in a total exposure of up to 48 weeks. The results at Week 48 were generally consistent with those at Week 16 and support sustained efficacy.
Study in complement inhibitor-naïve adult patients (APL2-308)
Study APL2-308 was an open-label, randomised, controlled study that enrolled patients with PNH who had not been treated with any complement inhibitor within 3 months prior to enrolment and with Hb levels less than the lower limit of normal (LLN). Eligible patients were randomised in a 2:1 ratio to receive pegcetacoplan or supportive care (e.g., transfusions, corticosteroids, supplements such as iron, folate, and vitamin B12), hereafter referred to as the control arm through the duration of the 26-week treatment period.to as the control arm through the duration of the 26-week treatment period.
Randomisation was stratified based on the number of PRBC transfusions within the 12 months prior to Day -28 (<4; ≥4). At any point during the study, a patient assigned to the control arm who had Hb levels ≥2 g/dL below baseline or presented with a PNH associated thromboembolic event was per protocol able to transition to pegcetacoplan for the remainder of the study.
A total of 53 patients were randomised, 35 to pegcetacoplan and 18 patients to the control arm. Demographics and baseline disease characteristics were generally well balanced between treatment arms. The mean age was 42.2 years in the pegcetacoplan arm and 49.1 years in the control arm. The mean number of PRBC transfusions in the 12 months prior to screening was 3.9 in the pegcetacoplan arm and 5.1 in the control arm. Five patients in each arm (14.3% in the pegcetacoplan arm and 27.8% in the control arm) had a history of aplastic anaemia. Further baseline values were as follows: mean baseline Hb levels (pegcetacoplan arm: 9.4 g/dL vs. control arm; 8.7 g/dL), ARC (pegcetacoplan arm: 230.2 × 109/L vs. control arm: 180.3 × 109/L), LDH (pegcetacoplan arm: 2 151.0 U/L vs. control arm: 1 945.9 U/L) and platelet count (pegcetacoplan arm: 191.4 × 109/L vs. control arm: 125.5 × 109/L). Eleven of 18 patients randomised to the control arm transitioned to pegcetacoplan because their Hb levels decreased by ≥2 g/dL below baseline. Of the 53 randomised patients, 52 (97.8%) received prophylactic antibiotic therapy according to local prescribing guidelines.
The primary and secondary efficacy endpoints were assessed at Week 26. The two co-primary efficacy endpoints were Hb stabilisation, defined as avoidance of a >1 g/dL decrease in Hb concentration from baseline in the absence of transfusion, and change in LDH concentration from baseline.
In the group treated with pegcetacoplan, 30 out of 35 patients (85.7%) achieved Hb stabilisation versus 0 patients in the control arm. The adjusted difference between pegcetacoplan and the control arm was 73.1% (95% CI, 57.2% to 89.0%; p<0.0001).
The least-square (LS) mean (SE) changes from baseline in LDH concentration at Week 26 were -1 870 U/L in the group treated with pegcetacoplan versus -400 U/L in the control arm (p<0.0001). The difference between pegcetacoplan and the control arm was -1 470 (95% CI, -2 113 to -827). Treatment differences between the pegcetacoplan and the control arm were evident at Week 2 and were maintained through Week 26 (Figure 3). LDH concentrations in the control arm remained elevated.
Figure 3. Mean (±SE) LDH concentration (U/L) over time by treatment group in study APL2-308:
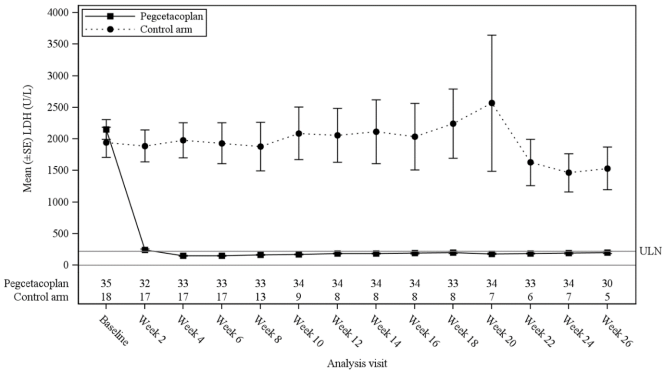
For the selected key secondary efficacy endpoints of haemoglobin response in the absence of transfusions, change in haemoglobin level, and change in ARC, the group treated with pegcetacoplan demonstrated a significant treatment difference versus the control arm (Table 3).
Table 3. Key secondary endpoints analysis in study APL2-308:
| Parameter | Pegcetacoplan (N=35) | Control arm (N=18) | Difference (95% CI) p-value |
|---|---|---|---|
| Haemoglobin response in the absence of transfusionsa n (%) | 25 (71%) | 1 (6%) | 54% (34%, 74%) p<0.0001 |
| Change from baseline to Week 26 in haemoglobin level (g/dL) LS Mean (SE) | 2.9 (0.38) | 0.3 (0.76) | 2.7 (1.0, 4.4) |
| Change from baseline to Week 26 in ARC (109/L) LS Mean (SE) | -123 (9.2) | -19 (25.2) | -104 (-159, -49) |
a Haemoglobin response was defined as a ≥1 g/dL increase in haemoglobin from baseline at Week 26.
ARC = Absolute reticulocyte count, CI = Confidence interval, LS = Least square, SE = Standard error
C3G and primary IC-MPGN
The efficacy and safety of pegcetacoplan in patients with C3G or primary IC-MPGN was assessed in the randomised, placebo-controlled, double- blinded phase 3 Study APL2-C3G-310, including adults and adolescents with native kidney or post-transplant recurrent C3G or primary IC-MPGN.
The dose of pegcetacoplan was 1 080 mg twice weekly for adults or adolescents with body weights ≥50 kg, or weight-based for adolescents with body weights <50 kg.
Study in adult and adolescent patients with C3G or primary IC-MPGN (APL2-C3G-310)
Study APL2-C3G-310 was a randomised, double-blinded study with a placebo-controlled period of 26-weeks, followed by a 26-week OLP. This study enrolled adolescents from 12 years to 17 years of age, and adults with C3G or primary IC-MPGN. This study enrolled patients with native kidney or post-transplant recurrent disease who presented with proteinuria ≥1 g/day and eGFR ≥30 mL/min/1.73 m². Patients were on a stable and optimised dose regimen for C3G/primary 16 IC-MPGN treatment (e.g., RAS inhibitors, sodium-glucose cotransporter-2 [SGLT-2] inhibitors, immunosuppressants, systemic corticosteroids no higher than 20 mg/day of prednisone equivalent) for at least 12 weeks prior to randomisation.
Eligible patients were randomised in a 1:1 ratio to receive pegcetacoplan or placebo subcutaneously twice weekly during the 26-week RCP. Two stratification factors were applied to the randomisation; patients with post-transplant recurrence versus native kidney disease patients, and patients with baseline renal biopsies (either collected during screening or within 28 weeks prior to randomisation) versus patients without baseline renal biopsies. During the RCP, changes to the baseline treatment regimens for C3G/primary IC-MPGN were minimised and only made when required for the well-being of the patient. Patients who completed the RCP, entered the 26-week OLP, in which all participants were treated with pegcetacoplan twice weekly.
A total of 124 patients were randomised, 63 to pegcetacoplan and 61 to placebo. Demographics and baseline disease characteristics were generally balanced between the two groups (see Table 4). A total of 118 patients completed the 26-week RCP, of which 114 patients completed the OLP treatment period with pegcetacoplan (N=59 pegcetacoplan-to-pegcetacoplan; N=55 placebo-to-pegcetacoplan).
Table 4. Patient baseline demographics and disease characteristics in study APL2-C3G-310:
| Parameter | Statistics | Pegcetacoplan (N=63) | Placebo (N=61) |
| Age (years) | Mean (SD) | 28.2 (17.1) | 23.6 (14.3) |
| Adolescents (12 – 17 years) | n (%) | 28 (44.4) | 27 (44.3) |
| Adults ≥18 years | n (%) | 35 (55.6) | 34 (55.7) |
| Sex | |||
| Male | n (%) | 26 (41.3) | 28 (45.9) |
| Female | n (%) | 37 (58.7) | 33 (54.1) |
| Type of disease at Screening | |||
| C3G | n (%) | 51 (81.0) | 45 (73.8) |
| C3GN | n (%) | 45 (71.4) | 41 (67.2) |
| DDD | n (%) | 4 (6.3) | 4 (6.6) |
| Undetermined | n (%) | 2 (3.2) | 0 |
| IC-MPGN | n (%) | 12 (19.0) | 16 (26.2) |
| Time since diagnosis of C3G/IC-MPGN (years) | Mean (SD) | 3.64 (3.47) | 3.76 (3.62) |
| Prior kidney transplant | n (%) | 5 (7.9) | 4 (6.6) |
| Time since last kidney transplant (years) | Mean (SD) | 11.4 (6.7) | 5.8 (6.4) |
| Time since most recent post-transplant recurrence (years) | Mean (SD) | 1.47 (1.49) | 1.38 (1.64) |
| Baseline triplicate FMU uPCR (mg/g) | Mean (SD) | 3124 (2408) | 2541 (2015) |
| Baseline eGFR (mL/min/1.73 m²) | Mean (SD) | 78.5 (34.1) | 87.2 (37.2) |
| C3c staining in baseline biopsy | |||
| 3+ | n (%) | 51 (81.0) | 51 (83.6) |
| 2+ | n (%) | 12 (19.0) | 10 (16.4) |
| Baseline serum albumin (g/dL) | Mean (SD) | 3.31 (0.61) | 3.39 (0.70) |
| Baseline serum C3 (mg/dL) | Mean (SD) | 60.6 (45.7) | 56.3 (35.6) |
| Disease manifestations | |||
| Oedema | n (%) | 45 (71.4) | 32 (52.5) |
| Fatigue | n (%) | 16 (25.4) | 8 (13.1) |
| Haematuria | n (%) | 37 (58.7) | 39 (63.9) |
| High Blood Pressure | n (%) | 35 (55.6) | 29 (47.5) |
| Nephrotic Syndrome | n (%) | 32 (50.8) | 27 (44.3) |
| Use of other treatments at baseline* | |||
| Agents acting on the renin-angiotensin system | n (%) | 59 (93.7) | 54 (88.5) |
| Immunosuppressants | n (%) | 49 (77.8) | 45 (73.8) |
| Glucocorticoids | n (%) | 29 (46.0) | 27 (44.3) |
* Within 12 weeks prior to study entry.
C3G = C3 glomerulopathy, C3GN = C3 glomerulonephritis, DDD = Dense-deposit disease, IC MPGN = Immune-complex membranoproliferative glomerulonephritis, FMU = First-morning urine, uPCR = Urine protein-to-creatinine ratio, eGFR = Estimated glomerular filtration rate, SD = Standard deviation
The primary and key secondary efficacy endpoints were assessed at Week 26. The primary efficacy endpoint was the log-transformed ratio of first-morning urine (FMU) uPCR at Week 26 compared with baseline.
Pegcetacoplan was superior to placebo, with a statistically significant 68.1% reduction (95% CI: 57.3% to 76.2%, p<0.0001) in uPCR from baseline compared to placebo after 26 weeks of treatment (-67.2% [95% CI: -74.9% to -57.2%] and +2.9% [95% CI: -8.6% to 15.9%] for pegcetacoplan and placebo respectively. Efficacy of similar magnitude was observed in subgroups irrespective of age (adolescents vs. adults), disease type (C3G vs. primary IC-MPGN), disease status (native vs. post-transplant recurrent disease), and concomitant use of immunosuppressants/glucocorticoids (yes vs. no). The effect of pegcetacoplan on uPCR was sustained through Week 52 (-67.2% from baseline). Patients who switched from placebo to pegcetacoplan at Week 26 (Figure 4) experienced a similar reduction (-51.3%) at Week 52.
Figure 4. Geometric mean ratio (95% CI) of FMU uPCR compared to baseline over time by treatment group from MMRM model in Study APL2-C3G-310:
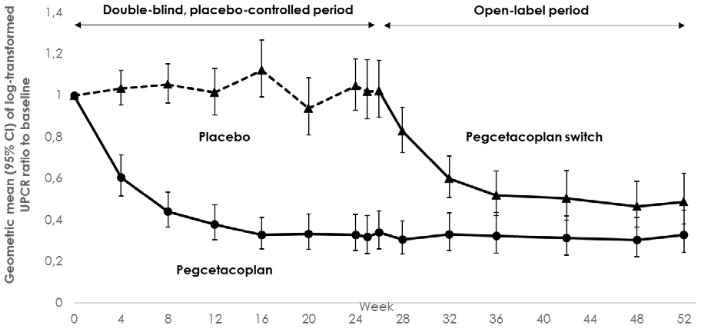
Note: Geometric mean ratio calculated from re-exponentiated LS Means.
CI = Confidence interval, LS = Least square, FMU = First-morning urine, uPCR = Urine protein-to-creatinine ratio, MMRM = Mixed model of repeated measure
Pegcetacoplan treatment for 26 weeks demonstrated statistically significant improvement in the key secondary endpoint related to proteinuria reduction, with 60.3% of patients treated with pegcetacoplan achieving a ≥50% reduction in uPCR compared to 4.9% in the placebo group, a difference of 52.7% (95% CI: 29.2%–76.2%; p<0.0001).
Pegcetacoplan treatment for 26 weeks resulted in a higher proportion of patients achieving a reduction of two orders of magnitude or greater, on a scale of 0-3, in renal C3 staining intensity with 26 (74.3%) patients on pegcetacoplan vs. 4 (11.8%) on placebo and a difference of 64.3% (95% CI: 41.4% -87.2%, nominal p<0.0001).
Pegcetacoplan treatment for 26 weeks showed stabilisation in eGFR with a change from baseline of -1.497 (2.242) on pegcetacoplan vs. -7.808 (1.919) on placebo, and a treatment difference of 6.312 mL/min/1.73m 2 (95% CI: 0.501, 12.122, nominal p=0.0333). The effect of pegcetacoplan on eGFR was sustained through Week 52. Patients who switched from placebo to pegcetacoplan at Week 26 experienced a similar stabilisation at Week 52.
Efficacy of similar magnitude was broadly observed for proteinuria reduction ≥50%, C3 staining clearance and eGFR stabilisation in subgroups irrespective of age (adolescents vs. adults), disease type (C3G vs. primary IC-MPGN), disease status (native vs. post-transplant recurrent disease) and concomitant use of immunosuppressants/glucocorticoids (yes vs. no) at Week 26.
Study in adult post-transplant recurrent C3G or primary IC-MPGN (APL2-C3G-204)
Study APL2-C3G-204 was a phase 2 open-label, randomised study in 13 adult patients with post-transplant recurrent C3G (N=10) or primary IC-MPGN (N=3) for 52 weeks.
During the first 12 weeks of the study, 10 patients received pegcetacoplan, in addition to standard of care (SOC), and 3 only SOC. All patients received pegcetacoplan from Week 13 to Week 52.
The primary endpoint of reduction in C3 staining intensity on renal biopsy at Week 12 was observed in 50% of the patients treated with pegcetacoplan (5 of 10 patients; 4 of which, achieved a staining score of zero), and 33.3% of the patients in the control group (1 of 3 patients; with this patient achieving a staining score of 1).
In general, changes and percentage changes from baseline in eGFR (secondary endpoint) were small. Mean (SD) eGFR changed from 52.3 (12.11) mL/min/1.73 m² at baseline to 57.3 (25.12) mL/min/1.73 m² at Week 52, and median eGFR changed from 50.5 mL/min/1.73 m² at baseline to 58.5 mL/min/1.73 m² at Week 52. Most patients (9 of 13 patients [69.2%]) across groups achieved stabilisation or improvement in eGFR by Week 52.
Immunogenicity
Two different assays for the detection of anti-pegcetacoplan peptide anti-drug antibody (ADA) were used in PNH and C3G or primary IC-MPGN clinical studies, respectively. The assay used for C3G or primary IC-MPGN was more sensitive. Differences in assays preclude meaningful comparisons of the incidence of ADAs in the studies described below.
In PNH clinical studies, ADA incidence (treatment-emergent ADA or boosted ADA from pre-existing level) was low, and when present, had no noticeable impact on the PK/PD, efficacy, or safety profile of pegcetacoplan. Throughout studies APL2-302 and APL2-308, 3 out of 126 patients who were exposed to pegcetacoplan had confirmed positive anti-pegcetacoplan peptide antibodies. All 3 patients also tested positive for neutralising antibody (NAb). NAb response had no apparent impact on PK or clinical efficacy. Eighteen out of 126 patients developed anti-PEG antibodies; 9 were treatment-emergent and 9 were treatment-boosted.
In C3G and primary IC-MPGN clinical studies, ADA incidence (treatment-emergent ADA or boosted ADA from pre-existing level) in study APL2-C3G-310 was 23.6% for anti-PEG and 16.3% for anti-pegcetacoplan peptide. Based on population PK and PD analysis, ADAs had no clinically meaningful impact on efficacy or PK/PD in a pooled analysis population. Five patients also tested positive for NAb. NAb response had no apparent impact on PK or clinical efficacy. Twenty-nine out of 123 patients developed anti-PEG antibodies;14 were treatment-emergent and 15 were treatment-boosted. In patients with post-transplant recurrent disease in study APL2-C3G-204, no patient developed a positive ADA response (treatment-emergent ADA or boosted ADA from pre-existing level) to pegcetacoplan peptide or PEG. During the 26-week placebo-controlled period in study APL2-C3G-310, there was no detectable impact of ADAs on the safety of pegcetacoplan treatment.
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with ASPAVELI in one or more subsets of the paediatric population in PNH and C3G or primary IC-MPGN, respectively (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Absorption
Pegcetacoplan is administered by subcutaneous infusion and gradually absorbed into the systemic circulation with a median Tmax between 108 and 144 hours (4.5 to 6.0 days) following a single subcutaneous dose to healthy volunteers.
Steady-state serum concentrations following twice weekly dosing at 1 080 mg in patients with PNH were achieved approximately 4 to 6 weeks following the first dose. In complement inhibitor-experienced patients (Study APL2-302) the geometric mean (%CV) steady-state serum concentrations ranged between 655 (18.6%) and 706 (15.1%) μg/mL in patients treated for 16 weeks. Steady-state concentrations in the patients (n=22) that continued to receive pegcetacoplan up to Week 48 were 623 μg/mL (39.7%), indicating sustainable therapeutic concentrations of pegcetacoplan through Week 48. In complement inhibitor-naïve patients (Study APL2-308) the geometric mean (%CV) steady-state serum concentration at Week 26 was 744 μg/mL (25.5%) with twice weekly dosing. The bioavailability of a subcutaneous dose of pegcetacoplan is estimated to be 76% based on population PK analysis.
Steady-state serum concentrations following twice weekly dosing at 1 080 mg in C3G or primary IC-MPGN patients were achieved approximately 4 to 8 weeks following the first dose and therapeutic concentrations of pegcetacoplan were maintained through Week 52. In patients of study APL2-C3G-310, the steady-state mean (%CV) serum concentrations ranged between 715.8 (31.2%) and 765.7 (23.2%) μg/mL up to Week 26 and remained between 670.1 (30.1%) and 726.6 (30.5%) μg/mL up to Week 52.
Distribution
The mean (%CV) volume of distribution of pegcetacoplan is approximately 3.98 L (32%) in patients with PNH based on population PK analysis.
The mean (%CV) of central volume of distribution of pegcetacoplan is approximately 4.31 L (32.1%) in adult patients with C3G or primary IC-MPGN.
Metabolism/elimination
Based on its PEGylated peptide structure, the metabolism of pegcetacoplan is expected to occur via catabolic pathways and be degraded into small peptides, amino acids, and PEG. Results of a radiolabelled study in cynomolgus monkeys suggest the primary route of elimination of the labelled peptide moiety is via urinary excretion. Although the elimination of PEG was not studied, it is known to undergo renal excretion.
Pegcetacoplan showed no inhibition or induction of the CYP enzyme isoforms tested as demonstrated from the results of in vitro studies. Pegcetacoplan was neither a substrate nor an inhibitor of the human uptake or efflux transporters.
Following multiple subcutaneous dosing of pegcetacoplan in patients with PNH, the mean (%CV) clearance is 0.015 L/h (30%) and median effective half-life of elimination (t1/2) is 8.6 days as estimated by the population PK analysis.
The estimated mean (CV%) of clearance is 0.012 L/hour (43%) in adult patients with C3G or primary IC-MPGN. The median terminal t½ is 10.1 days in adult patients with C3G or primary IC-MPGN.
Linearity/non-linearity
Exposure of pegcetacoplan increases in a dose proportional manner from 45 to 1 440 mg.
Special populations
No impact on the pharmacokinetics of pegcetacoplan was identified with age (12-81 years), race or sex based on the results of population PK analysis in patients with PNH, C3G or primary IC-MPGN.
Compared with a reference 70 kg patient, the steady-state average concentration is predicted to be approximately 20% higher in patients with a body weight of 50 kg. PNH patients weighing 40 kg are predicted to have a 45% higher average concentration. Minimal data are available on the safety profile of pegcetacoplan for PNH patients with a body weight below 50 kg.
Elderly
Although there were no apparent age-related differences observed in these studies, the number of patients aged 65 years and over is not sufficient to determine whether they respond differently from younger patients. See section 4.2.
Paediatric population
Based on population PK analysis, body weight in adolescent patients (12-17 years) has an impact on clearance and volume of distribution. The dosing regimen for adolescents with C3G or primary IC-MPGN is based on the patient´s body weight. See section 4.2. The model-predicted exposure for adolescents with C3G or primary IC-MPGN is adequately matched to the adult reference exposure.
Renal impairment
In a study of 8 patients with severe renal impairment, defined as creatinine clearance (CrCl) less than 30 mL/min using the Cockcroft-Gault formula (with 4 patients with values less than 20 mL/min), renal impairment had no effect on the pharmacokinetics of a single 270-mg dose of pegcetacoplan. There are minimal data on patients with PNH with renal impairment who have been administered the clinical dose of 1 080 mg twice weekly. Based on population PK analysis, eGFR had no clinically meaningful impact on pegcetacoplan exposure in a pooled analysis population. There are no available clinical data for the use of pegcetacoplan in patients with ESRD requiring dialysis. See section 4.2.
5.3. Preclinical safety data
In vitro and in vivo toxicology data reveal no toxicity of special concern for humans. Effects observed in animals at exposure levels similar to clinical exposure levels are described below. These effects were not observed in clinical studies.
Animal reproduction
Pegcetacoplan treatment of pregnant cynomolgus monkeys at a subcutaneous dose of 28 mg/kg/day (2.9 times the human steady-state Cmax) from the gestation period through parturition resulted in a statistically significant increase in abortions or stillbirths. No maternal toxicity or teratogenic effects were observed in offspring delivered at term. Additionally, no developmental effects were observed in infants up to 6 months postpartum. Systemic exposure to pegcetacoplan was detected in foetuses from monkeys treated with 28 mg/kg/day from the period of organogenesis through the second trimester, but the exposure was minimal (less than 1%, not pharmacologically significant).
Carcinogenesis
Long term animal carcinogenicity studies of pegcetacoplan have not been conducted.
Genotoxicity
Pegcetacoplan was not mutagenic when tested in in vitro bacterial reverse mutation (Ames) assays and was not genotoxic in an in vitro assay in human TK6 cells or in an in vivo micronucleus assay in mice.
Animal toxicology
Repeat-dose studies were conducted in rabbits and cynomolgus monkeys with daily subcutaneous doses of pegcetacoplan up to 7 times the human dose (1 080 mg twice weekly). Histologic findings in both species included dose-dependent epithelial vacuolation and infiltrates of vacuolated macrophages in multiple tissues. These findings have been associated with large cumulative doses of long-chain PEG in other marketed PEGylated drugs, were without clinical consequence, and were not considered adverse. Reversibility was not demonstrated in the pegcetacoplan animal studies after one month and was not evaluated for a longer duration. Data from literature suggest reversibility of PEG vacuoles.
Renal tubular degeneration was observed microscopically in both species at exposures (Cmax and AUC) less than or comparable to those for the human dose and was minimal and nonprogressive between 4 weeks and 9 months of daily administration of pegcetacoplan. Although no overt signs of renal dysfunction were observed in animals, the clinical significance and functional consequence of these findings are unknown.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.