AUSTEDO Prolonged-release tablet Ref.[116189] Active ingredients: Deutetrabenazine
Source: European Medicines Agency (EU) Revision Year: 2026 Publisher: TEVA GmbH, Graf-Arco-Str. 3, 89079 Ulm, Germany
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Other nervous system drugs
ATC code: N07XX16
Mechanism of action
Deutetrabenazine and the major circulating metabolites (deuterated α-HTBZ and deuterated β-HTBZ), are reversible inhibitors of VMAT2, resulting in decreased uptake of monoamines into synaptic vesicles and depletion of monoamine stores in dopaminergic regions (e.g. striatum and cortex) of the brain (see section 5.2 "Distribution"). While the precise mechanism of action by which deutetrabenazine exerts its effects in the treatment of tardive dyskinesia is unknown, it is believed to be related to its effect as a depletor of monoamines (such as dopamine, serotonin, norepinephrine, and histamine) from nerve terminals.
Pharmacodynamic effects
Cardiac electrophysiology
At the maximum recommended dose, deutetrabenazine does not prolong the QTc interval to any clinically relevant extent. An exposure-response analysis on QTc prolongation from a study conducted in extensive, intermediate and poor CYP2D6 metabolisers, showed that a clinically relevant effect can be excluded at exposures following daily doses of 24 and 48 mg of deutetrabenazine.
Clinical efficacy and safety
The efficacy of deutetrabenazine was assessed in two 12-week, randomised, double-blind, placebo-controlled trials in adult patients with tardive dyskinesia presenting with symptoms that were bothersome to the patient or caused functional impairment (n=335). These studies included patients who had moderate or severe abnormal movements based on Item 8 of the Abnormal Involuntary Movement Scale (AIMS) and a total motor AIMS score of ≥6 (based on Items 1 through 7). Enrolled patients had a history of using a dopamine receptor antagonist (DRA, e.g. antipsychotics, metoclopramide) for at least 3 months (or 1 month in patients 60 years of age and older), were psychiatrically stable and had no change in psychoactive medications for at least 30 days (45 days for antidepressants). Background comorbid illnesses included schizophrenia/schizoaffective disorder (n=207, 62%), mood disorder (n=112, 33%), other (neurological, psychiatric and gastrointestinal conditions; n=15, 4%), and missing (n=1, <1%). With respect to concurrent DRA use, 75.5% of the patients were on a stable DRA dose, while 24.5% were not receiving a DRA at baseline. The primary efficacy endpoint in the trials was the total motor AIMS score (the sum of Items 1 to 7 with score range from 0 to 28).
Fixed-dose study (AIM-TD - Study 1)
Study 1 was a 12-week, double-blind, placebo-controlled, fixed-dose trial in adult patients with tardive dyskinesia. A total of 222 patients were randomised into one of four arms: 12 mg deutetrabenazine per day, 24 mg deutetrabenazine per day, 36 mg deutetrabenazine per day, or placebo administered orally. The study included a 4-week dose escalation period and an 8-week maintenance period. The dose of deutetrabenazine was started at 12 mg per day and increased at weekly intervals in 6 mg per day increments to the targeted fixed doses of 12 mg, 24 mg or 36 mg deutetrabenazine per day.
Demographics and baseline disease characteristics were comparable between the study arms. Patients had a mean age of 57 years (range: 21 to 81 years), 24% were 65 years of age and older, 48% were male, and 79% were Caucasian.
Deutetrabenazine showed a statistically significant and clinically meaningful improvement in the AIMS total score from baseline compared to placebo for the 24 mg and 36 mg arms (see Table 2). The effect occurred from as early as week 2 and was sustained over the treatment period (see Figure 1).
Table 2. Improvement in AIMS total score in Study 1:
| Efficacy endpoint | Placebo (n=58) | Deutetrabenazine 12 mg/day (n=60) | Deutetrabenazine 24 mg/day (n=49) | Deutetrabenazine 36 mg/day (n=55) |
| AIMS total score | ||||
| Mean baseline score (SD) | 9.5 (2.71) | 9.6 (2.40) | 9.4 (2.93) | 10.1 (3.21) |
| LS Mean change from baseline (SE) | -1.4 (0.41) | -2.1 (0.42) | -3.2 (0.45) | -3.3 (0.42) |
| Treatment effect (95% CI) | -0.7 (-1.84, 0.42) | -1.8 (-3.00, -0.63) | -1.9 (-3.09, -0.79) | |
| p-value | 0.001* | |||
LS Mean = Least-squares mean; SD = Standard deviation; SE = Standard error; CI = 2-sided 95% confidence interval
* Multiplicity-adjusted p-value for difference from placebo, statistically significant
Figure 1. Mean change from baseline in AIMS total score in Study 1:
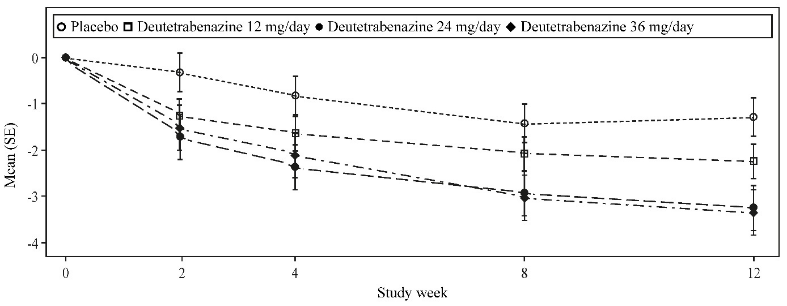
Flexible-dose study (ARM-TD - Study 2)
Study 2 was a 12-week, double-blind placebo-controlled, flexible-dose trial in adults with tardive dyskinesia. A total of 113 patients received daily doses of placebo or deutetrabenazine, starting at 12 mg per day with increases allowed at weekly intervals in 6 mg per day increments until adequate dyskinesia control was achieved, a clinically significant adverse reaction occurred, or the maximum daily dose of 48 mg deutetrabenazine per day was reached. The study included a 6-week dose titration period and a 6-week maintenance period. Patients had a mean age of 55 years (range: 25 to 75 years), 14% were 65 years of age and older, 48% were male, and 70% were Caucasian.
The average dose of deutetrabenazine at the end of treatment was 38.3 mg per day. Deutetrabenazine showed a statistically significant and clinically meaningful improvement in the AIMS total score from baseline compared to placebo (see Table 3).
Table 3. Improvement in AIMS total score in Study 2:
| Efficacy endpoint | Placebo (n=57) | Deutetrabenazine 12 mg/day – 48 mg/day (n=56) |
| AIMS total score | ||
| Mean baseline score (SD) | 9.6 (3.78) | 9.7 (4.14) |
| LS Mean change from baseline (SE) | -1.6 (0.46) | -3.0 (0.45) |
| Treatment effect (95% CI) | -1.4 (-2.6, -0.2) | |
| p-value | 0.0188* | |
LS Mean = Least-squares mean; SD = Standard deviation; SE = Standard error; CI = 2-sided 95% confidence interval
* Multiplicity-adjusted p value for difference from placebo, statistically significant
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with deutetrabenazine in all subsets of the paediatric population in treatment of tardive dyskinesia (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
After oral dosing deutetrabenazine undergoes rapid and extensive hepatic metabolism to its active metabolites deuterated α-HTBZ and deuterated β-HTBZ resulting in low plasma concentrations of deutetrabenazine compared to that of the active metabolites.
Absorption
Following oral administration of deutetrabenazine, the extent of absorption is at least 80%. Peak plasma concentrations of deutetrabenazine and its active metabolites (deuterated α-HTBZ and deuterated β-HTBZ) are reached within 3 hours after repeated dosing, followed by sustained plateaus for several hours allowing for a 24-hour dosing interval. Absorption is not influenced by food intake.
Distribution
The protein binding of deutetrabenazine, deuterated α-HTBZ and deuterated β-HTBZ in human plasma is 82%, 57%, and 49% respectively, with no preferential binding of total radioactivity to the cellular components of human blood after 14C-deutetrabenazine administration.
Single oral dose of either 14C-deutetrabenazine or 14C-tetrabenazine to rats in a quantitative whole-body autoradiography study resulted in similar blood to brain ratios. Results of PET-scan studies in humans showed that following intravenous injection of 11C-tetrabenazine or α-HTBZ, radioactivity is rapidly distributed to the brain, with the highest binding in the striatum and lowest binding in the cortex. No human PET-scan studies have been performed with deutetrabenazine.
Based on population pharmacokinetic modelling, after oral administration, the apparent volumes of distribution (Vc/F) for deutetrabenazine, deuterated α-HTBZ, and deuterated β-HTBZ is 13 700 L, 490 L, and 860 L, respectively.
Biotransformation
In vitro studies using human liver microsomes demonstrate that deutetrabenazine is extensively biotransformed, mainly by carbonyl reductase, to its major active metabolites, deuterated α-HTBZ and deuterated β-HTBZ, which are subsequently metabolised primarily by CYP2D6, with minor contributions of CYP1A2 and CYP3A4/5, to form several minor metabolites.
Deutetrabenazine and its active metabolites did not inhibit or induce any CYP enzymes that were studied in vitro at clinically relevant concentrations.
Elimination
In a mass balance study in six healthy subjects, 75% to 86% of the deutetrabenazine dose was excreted in the urine, and faecal recovery accounted for 8% to 11% of the dose. Urinary excretion of deuterated α-HTBZ and deuterated β-HTBZ each accounted for less than 10% of the administered dose. Sulphate and glucuronide conjugates of deuterated α-HTBZ and deuterated β-HTBZ, as well as products of oxidative metabolism, accounted for the majority of metabolites in the urine.
Based on population pharmacokinetic modelling, after oral administration, for deutetrabenazine, deuterated α-HTBZ and deuterated β-HTBZ, the apparent clearance values (CL/F) are 11,750 L/h, 67 L/h, and 260 L/h; the half-lives are 11.4 h, 11 h, and 8.2 h.
Deutetrabenazine and its active metabolites are not substrates or inhibitors of the human transporters, predominantly located in the liver, intestines, central nervous system (CNS) and kidney, that were studied in vitro at clinically relevant concentrations.
Linearity/non-linearity
Dose proportionality was observed in the dose range of 12 mg to 48 mg.
Special populations
Based on population pharmacokinetic analyses there is no apparent effect of gender, race, and age (18-64 years) on the pharmacokinetics of deuterated α-HTBZ and deuterated β-HTBZ.
Limited pharmacokinetic data are available for patients 65-74 years of age (approx. 9% of the patients) and 75-84 years of age (approx. 1% of the patients). No data are available for those over 85 years of age. Therefore, no definitive pharmacokinetic conclusions can be made for patients over 65 years of age.
The majority of patients had a body weight of 50 kg to <120 kg, and only a limited number of patients with a body weight of <50 kg or ≥120 kg were included in clinical trials. Population pharmacokinetic analyses predict higher exposures of deuterated α-HTBZ and deuterated β-HTBZ in patients with lower body weights and lower exposures in patients with higher body weights, however, body weight was not correlated to individual response as measured by change in AIMS total score at week 12 of treatment.
Renal impairment
No clinical studies have been conducted to assess the effect of renal impairment on the pharmacokinetics of deuterated α-HTBZ and deuterated β-HTBZ. Based on population pharmacokinetic analyses, the effect of renal impairment on the pharmacokinetic exposures of deuterated total (α+β)-HTBZ is negligible. As the major route of elimination of the active metabolites is non-renal, it is unlikely that patients with any degree of renal impairment will be exposed to excessive concentrations of deutetrabenazine and its active metabolites.
Hepatic impairment
The effect of hepatic impairment on the pharmacokinetics of deutetrabenazine and its active metabolites has not been studied. Since deutetrabenazine is extensively metabolised in the liver and due to the potential increase in systemic exposure the use of deutetrabenazine in patients with hepatic impairment is contraindicated (see section 4.3).
Poor CYP2D6 metabolisers
Although the pharmacokinetics of deutetrabenazine and its metabolites have not been systematically evaluated in patients who do not express the drug-metabolising enzyme CYP2D6, data in healthy subjects who are poor CYP2D6 metabolisers shows that the exposure to deuterated α-HTBZ and deuterated β-HTBZ would be increased similarly to taking strong CYP2D6 inhibitors (maximum exposure increased 2-fold and total exposure approximately 4-fold) (see sections 4.2 and 4.5).
5.3. Preclinical safety data
After 4 weeks of dosing (interim necropsy) in a 3-month toxicology study of deutetrabenazine in rats, observations of oestrus cycle arrest at the pro-oestrus (pre-ovulatory) phase and mammary hyperplasia in females at exposures similar to those expected in patients were likely physiological consequences of reduced CNS dopamine with attendant disinhibition of prolactin. CNS-related observations in this study, including intermittent tremors, partial eye closure, changes in activity, and twitching ears, that were observed at clinically relevant doses, with subclinical exposure to some major metabolites, were likely associated with depletion of monoamine neurotransmitter stores. Male rats from the 3-month study, at deutetrabenazine exposures slightly below clinical exposure levels, showed adverse effect of decreased body weight gains.
Non-rodent toxicology studies of deutetrabenazine were not conducted. However, observations in a toxicology study of another VMAT2 inhibitor (tetrabenazine) that was orally administered to dogs for 9 months revealed CNS-related pharmacological effects similar to rats, including hypoactivity, lethargy, strabismus, or closed eyes at doses associated with major human metabolite exposures below clinical exposure levels.
Deutetrabenazine and its major active metabolites, deuterated α-HTBZ and deuterated β-HTBZ, were not genotoxic in a standard battery of in vitro assays, and deutetrabenazine treatment resulted in a negative response for the induction of bone marrow micronuclei in mice.
Carcinogenicity studies of deutetrabenazine were not conducted. Deutetrabenazine had no effect on embryofoetal development when administered to pregnant rats at doses up to 30 mg/kg/day corresponding to exposure levels (AUC) 119-fold higher than in patients at the maximum recommended dose. Exposure levels (AUC) to deuterated α-HTBZ were comparable and deuterated β-HTBZ metabolites were slightly lower than the metabolite levels in humans at the maximum recommended dose.
No embryofoetal development studies in a non-rodent species, or fertility and pre- and post-natal development studies in a rodent species were conducted with deutetrabenazine.
Tetrabenazine had no effects on embryofoetal development when administered to pregnant rabbits during the period of organogenesis at oral doses up to 60 mg/kg/day.
When tetrabenazine was orally administered to pregnant rats (5, 15, and 30 mg/kg/day) from the beginning of organogenesis through the lactation period, an increase in stillbirths and offspring postnatal mortality was observed at 15 and 30 mg/kg/day and delayed pup maturation was observed at all doses. A no-effect dose for pre- and postnatal developmental toxicity in rats was not identified.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.