BRINSUPRI Film-coated tablet Ref.[116035] Active ingredients: Brensocatib
Source: European Medicines Agency (EU) Revision Year: 2025 Publisher: Insmed Netherlands B.V., Stadsplateau 7, 3521 AZ Utrecht, Netherlands
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: not yet assigned
ATC code: not yet assigned
Mechanism of action
Brensocatib is a competitive and reversible inhibitor of dipeptidyl peptidase 1 (DPP1). DPP1 activates pro-inflammatory neutrophil serine proteases (NSPs) during neutrophil maturation in the bone marrow. Brensocatib reduces the activity of NSPs implicated in the pathogenesis of bronchiectasis, including neutrophil elastase, cathepsin G and proteinase 3.
Pharmacodynamic effects
At 5 times the maximum recommended daily dose of brensocatib, there was no effect on QTc prolongation.
Clinical efficacy
The efficacy of brensocatib was assessed in a Phase 3, randomised, double-blind, placebo-controlled, parallel-group, multicentre, multinational trial (ASPEN) with a total of 1 721 patients 12 years of age and older with NCFB (1 680 adults and 41 adolescents).
All patients were randomised to one of two doses of brensocatib (25 mg: n=575; 10 mg: n=583) or placebo (n=563), administered once daily for 52 weeks.
All adult patients enrolled had a history of confirmed NCFB by chest computed tomography with at least 2 documented pulmonary exacerbations prior screening in the past 12 months. Adolescent patients had at least one pulmonary exacerbation in the prior 12 months. A qualifying exacerbation was defined by the need for a physician-prescribed course of systemic antibiotics for signs and symptoms of respiratory infection.
Demographics and baseline characteristics of ASPEN are provided in Table 2.
Table 2. Demographics and baseline characteristics of patients in ASPEN:
| ASPEN (N=1 721) | |
|---|---|
| Age (years), mean (SD) | 60 (16) |
| Female n (%) | 1 107 (64) |
| White n (%) | 1 266 (74) |
| Black or African American n (%) | 10 (1) |
| Asian n (%) | 191 (11) |
| Hispanic or Latino n (%) | 511 (30) |
| ≥3 PEx in prior 12 months n (%) | 502 (29) |
| Former smoker n (%) | 510 (30) |
| ppFEV1 post-bronchodilator, mean (SD) | 74 (23) |
| Sputum positive for Pseudomonas aeruginosa n (%) | 607 (35) |
| Chronic macrolide therapy n (%) | 329 (19) |
N = number of patients in the intent-to-treat analysis set; n = number of patients; PEx = pulmonary exacerbations; pp = percent predicted; FEV1 = forced expiratory volume in 1 second; SD = standard deviation
Exacerbations
The primary efficacy endpoint in ASPEN was the annualised rate of pulmonary exacerbations (PEx) over the 52-week treatment period.
Pulmonary exacerbations were defined as worsening of 3 or more major symptoms over 48 hours with increase in cough, sputum volume, sputum purulence or increased breathlessness or decreased exercise tolerance and fatigue and/or malaise and, haemoptysis, resulting in a healthcare provider's decision to prescribe systemic antibiotics. Pulmonary exacerbations were considered as severe if requiring treatment with intravenous antibiotics and/or resulted in hospitalisation.
In ASPEN, treatment with 25 mg of brensocatib in patients with NCFB demonstrated significant reductions in the annualised rate of pulmonary exacerbations compared with placebo. Key results are shown in Table 3.
Table 3. Exacerbations endpoints over 52 weeks in ASPEN:
| Brensocatib 25 mg (N=575) | Placebo (N=563) | ||
|---|---|---|---|
| Annualised rate of PEx | 1.04 | 1.29 | Rate ratio (95% CI): 0.81 (0.69, 0.94) |
| Median time to first PEx (weeks) | 50.71 | 36.71 | Hazard ratio (95% CI): 0.83 (0.70, 0.97) |
| Proportion of patients that were exacerbation free at week 52 (%) | 48.5 | 40.3 | Odds ratio (95% CI): 1.40 (1.10, 1.79) |
Lung function
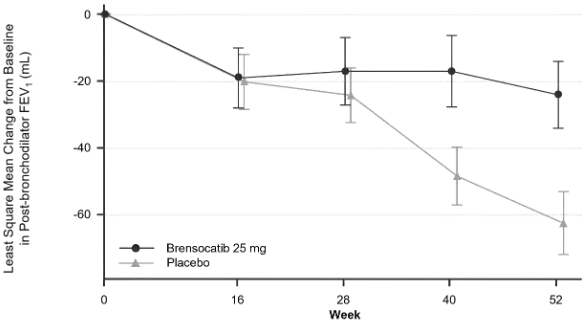
Change from baseline in post-bronchodilator FEV1 was assessed as a secondary endpoint. A dose of brensocatib 25 mg significantly reduced FEV1 decline in comparison to placebo at week 52 (Least Squares mean difference 38; 95% CI: 11, 65) (Figure 1).
Figure 1. LS mean change (SE) from baseline in post-bronchodilator FEV1 (mL) over time:
Paediatric population (adolescents)
In the pivotal 52-week study, 41 adolescents (12 to <18 years) were randomised to brensocatib 25 mg once daily, brensocatib 10 mg once daily or placebo. The adolescent subgroup was small and the study was not powered for efficacy in adolescents; confidence intervals were wide and results are inconclusive. Trends towards fewer pulmonary exacerbations and positive changes in post-bronchodilator FEV1 were observed with 25 mg brensocatib versus placebo. Safety and pharmacokinetic data in adolescents were generally consistent with adults (see sections 4.8 and 5.2).
The European Medicines Agency has deferred the obligation to submit the results of studies with Brinsupri in one or more subsets of the paediatric population in NCFB (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Absorption
The absolute oral bioavailability of brensocatib has not been studied in humans. Brensocatib is rapidly absorbed after oral administration. Tmax for tablets is approximately 1 hour in patients. Brensocatib oral absorption is not affected by food intake. Co-administration with a high fat meal delayed the time to reach peak concentration by 0.75 hours, however, the extent of brensocatib absorption remained the same.
Distribution
After oral administration, the volume of distribution at steady state was 126-138 L (CV: 22.4-23.3%) in adult patients and 71.3-83.6 L (CV: 19.9-26.3%) in adolescents with NCFB. The protein binding of brensocatib to human plasma was 82.2%-87.2%.
Biotransformation
Brensocatib undergoes metabolism primarily by CYP3A. Brensocatib accounted for 16.2% of the total radioactivity in plasma. Only one major circulating metabolite, thiocyanate, was detected in plasma. Thiocyanate is an endogenous compound, and clinical data showed that thiocyanate plasma concentrations were not affected and remained in the normal range on brensocatib treatment.
Interactions
In vitro studies
CYP450 enzymes:
Brensocatib is a substrate of CYP3A. Brensocatib does not inhibit CYP1A2, CYP2C8, CYP2C9, CYP2C19, or CYP2D6. In vitro studies are inconclusive regarding the potential of brensocatib to induce CYP2B6 and CYP3A4. In vivo induction cannot be excluded.
Transporter systems:
Brensocatib is a substrate of P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP). Brensocatib is not a substrate of MATE1, MATE2-K, OAT1, OAT3, OATP1B1, OATP1B3, OCT1 and OCT2. Brensocatib is not an inhibitor of P-gp, BCRP, OATP1B1, OATP1B3, OAT1, OAT3, OCT2, MATE1, and MATE2-K.
Effect of brensocatib on other medicinal products
In vitro data and population pharmacokinetic analyses indicate that brensocatib is unlikely to inhibit or significantly induce the activity of CYP isozymes or drug transporters at clinically relevant dose levels. However, in vitro studies were inconclusive regarding the potential of brensocatib to induce CYP2B6 and CYP3A4, and in vivo induction cannot be excluded.
Effect of other medicinal products on brensocatib
Brensocatib AUC and Cmax increased by 55% and 68% with a strong CYP3A inhibitor (e.g. clarithromycin) and by 32% and 53% with a strong P-gp inhibitor (e.g. verapamil) but decreased by 33% and 15% with a strong CYP3A inducer (e.g. rifampicin). Cmax and AUC remained unchanged with a potent proton-pump inhibitor (e.g. esomeprazole). The interaction effect on brensocatib systemic exposure is not clinically meaningful.
Elimination
Following a single oral dose of radiolabelled brensocatib, 54.2% of dose was excreted in urine and 28.3% in faeces with most radioactivity excreted within 72 hours. The unchanged brensocatib in urine and faeces were 22.8% and 2.41% of dose, respectively.
Terminal half-life was 32.6-39.6 hours (CV: 26.6-33.0%) in adult patients and 26.9-27.8 hours (CV: 26.8-37.3%) in adolescent patients.
Linearity/non-linearity
Brensocatib exhibits linear and time-independent pharmacokinetics with low to moderate intra- and inter-subject variability over a dose range of 5-120 mg following single administration and a dose range of 10-40 mg following once-daily administration. Population pharmacokinetics analysis using pooled data from 11 clinical studies in healthy subjects (n = 291) and patients with NCFB (n = 783) showed that brensocatib pharmacokinetics can be adequately described by a 2-disposition compartments with first-order oral absorption.
Pharmacokinetic/pharmacodynamic relationships
Exposure-response relationships were observed between brensocatib exposure (AUC) and clinical efficacy (i.e. decline of lung function measured as FEV1). At 25 mg, >99% NCFB patients in the ASPEN trial achieved an AUC threshold that was associated with clinically meaningful improvement in FEV1. No exposure-response relationships were detected for the occurrence of periodontal disease or pneumonia. A relationship between brensocatib exposure (AUC) and hyperkeratosis (mild and moderate) was observed. However, the predicted probability of mild or moderate hyperkeratosis was low at brensocatib 25 mg (3.01% in adults and 3.36% in adolescents).
Special populations
Population pharmacokinetic analysis showed no evidence of a clinically significant effect of age (range: 12 to 85 years), sex, race/ethnicity or body weight (range: 32 to 155 kg) on the pharmacokinetics of brensocatib.
Paediatric population
Based on the population pharmacokinetic analysis, there was no clinically meaningful age-related difference in the pharmacokinetics of brensocatib between adults and adolescents aged 12 to 17 years. Brensocatib has not been studied in children under 12 years of age (see section 4.2).
Hepatic impairment
In subjects with mild, moderate or severe hepatic impairment (Child-Pugh scores 5 to 12), brensocatib clearance following a single dose was comparable to that in healthy subjects. No dose adjustments are recommended for patients with mild, moderate or severe hepatic impairment (see section 4.2).
Renal impairment
In subjects with mild, moderate or severe renal impairment, (creatinine clearance ≥15 mL/min/1.73 m² and not requiring dialysis), brensocatib clearance following a single dose was comparable to that in healthy subjects. No dose adjustments are recommended for patients with mild, moderate or severe renal impairment (see section 4.2).
5.3. Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, genotoxicity and carcinogenic potential. Effects in non-clinical studies were observed only at exposures considered sufficiently in excess of the maximum human exposure indicating little relevance to clinical use.
General toxicity
In a 6-month rat study microscopic changes in the kidney (basophilic tubules in the outer medulla) and the lung (perivascular neutrophil infiltration and vacuolated macrophage accumulation consistent with phospholipids) were observed at 50 mg/kg/day. The no-observed-adverse-effect-level was considered to be 9 mg/kg/day (AUC 20 times the maximum recommended human dose [MRHD]).
In a 9-month dog study no adverse findings were observed at any dose (AUC 5 times the MRHD). In a preceding 6-month dog study, administration of brensocatib at 50 mg/kg/day caused periodontal disease resulting in early termination of the group. At ≥8 mg/kg/day, dose-dependent microscopic findings were noted in the testis (seminiferous tubule degeneration and atrophy), in the epididymis (decreased number of spermatozoa and cellular debris), and in the lung (accumulations of vacuolated macrophages consistent with phospholipids). At 50 mg/kg/day, additional microscopic findings were noted in the kidney (tubular regeneration) and in the lymphoid tissues (axillary, mandibular and mesenteric lymph nodes, gut associated lymphoid tissue and spleen) as indicated by the accumulations of vacuolated macrophages.
Reproductive and developmental toxicity
In a rat fertility and embryo-foetal development study, following treatment with brensocatib from 2 weeks prior to mating, during mating and up to the end of major embryonic organogenesis, recoverable minor malformations of bent scapula and wavy ribs were noted at plasma exposure (AUC) 128-times the human exposure at the MRHD. There was an increased incidence of skeletal variations (malpositioned pelvic girdle and vestigial supernumerary full and/or short ribs in both cervical and thoracolumbar regions) and differences in ossification at AUC ≥42-times the human exposure at the MRHD. The no effect dose for developmental toxicity was at AUC of 3-times the human exposure at the MRHD. In a rabbit embryo-foetal development study, treatment with brensocatib during implantation and major organogenesis induced maternal toxicity (reductions in body weight gain and food consumption) at AUC ≥5-times the human exposure at the MRHD. There were no adverse developmental effects at AUC 20-times the human exposure at the MRHD.
In a pre- and post-natal development study in rats treated from gestation day 6 through lactation day 20, no adverse findings were observed at any dose (up to AUC 17-times the human exposure at the MRHD). Brensocatib was detected in pups, suggesting that male and female pups were likely exposed via maternal milk during lactation.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.