BRINSUPRI Tablet Ref.[115811] Active ingredients: Brensocatib
Source: FDA, National Drug Code (US) Revision Year: 2025
12.1. Mechanism of Action
Brensocatib is a competitive, reversible inhibitor of dipeptidyl peptidase 1 (DPP1). DPP1 activates pro-inflammatory neutrophil serine proteases (NSPs) during neutrophil maturation in the bone marrow. Activated NSPs are implicated in the pathogenesis of neutrophil-mediated NCFB inflammation. In cell-based assays, DPP1 inhibition by brensocatib reduces the activity of NSPs including neutrophil elastase, cathepsin G, and proteinase 3.
12.2. Pharmacodynamics
Brensocatib prevents the activation of NSPs via DPP1 inhibition resulting in decreases in NSP activity in patients. In exploratory assays, brensocatib was associated with dose-dependent reductions in NSP activity in patients with NCFB.
Cardiac Electrophysiology
At concentrations approximately 2 times the steady state peak concentration of the maximum recommended daily dose of brensocatib in adult and adolescents with NCFB, clinically significant QTc interval prolongation was not observed.
12.3. Pharmacokinetics
Following once daily administration of brensocatib 10 mg, the estimated geometric mean (CV%) Cmax is 85.4 ng/mL (33%) and AUCtau is 1360 ng*h/mL (41%). Following once daily administration of brensocatib 25 mg, the estimated geometric mean (CV%) Cmax is 259 ng/mL (31%) and AUCtau is 4060 ng*h/mL (39%).
Brensocatib Cmax and AUCtau increase in a greater than dose proportional manner between doses of 10 mg and 1.6 times the highest recommended dose. Between doses of 10 mg and 1.6 times the highest recommended dose brensocatib once daily, brensocatib geometric mean steady state Cmax increased by 5.1-fold and AUCtau increased by 5.3-fold. In healthy subjects, at steady state, Cmax increased by about 1.5-fold and AUCtau increased by about 2-fold when compared to those observed following a single dose.
No clinically relevant differences in brensocatib pharmacokinetics were observed between healthy subjects and patients with NCFB.
Absorption
The absolute oral bioavailability of brensocatib has not been studied in humans. Based on data from a mass balance study in healthy subjects, the oral absorption of brensocatib in humans is greater than 80%. Following a single dose of 10 mg or 25 mg brensocatib, the median (min, max) time to maximum plasma concentration (Tmax) is 1.0-1.4 hours (0.5, 3.0 hours).
Effect of Food
No clinically relevant differences in brensocatib pharmacokinetics were observed following administration of brensocatib with a high-fat meal (990 calories, 52% fat).
Distribution
Following once daily administration of 10 mg or 25 mg brensocatib in patients with NCFB, the estimated volume of distribution at steady state ranged from 126 to 138 L. The protein binding of brensocatib in human plasma was 87.2%.
Elimination
Following a single oral administration of brensocatib at 10 mg or 25 mg in healthy subjects, the elimination half-life ranged from 25 to 39 hours and the apparent oral clearance ranged from 6.4 to 10.7 L/hour.
Metabolism
Brensocatib is primarily metabolized by CYP3A and to a lesser extent by CYP2C8 and CYP2D6. Brensocatib accounted for 16.2% of the total radioactivity in plasma. One major circulating metabolite, thiocyanate, was identified in plasma and accounted for 51% of the total radioactivity AUC following administration of a radio-labeled brensocatib dose. In patients with NCFB at recommended doses of brensocatib, no clinically meaningful changes of plasma thiocyanate from baseline were observed.
Excretion
Following administration of a single oral dose of radiolabeled brensocatib 1.6 times the highest recommended dose to healthy subjects, 54.2% of dose was recovered in urine (22.8% as unchanged brensocatib) and 28.3% of dose was recovered in feces (2.4% as unchanged brensocatib).
Specific Populations
No clinically significant differences in the pharmacokinetics of brensocatib were observed based on age (12 to 85 years), sex, race (72% White, 6% Black, and 12% Asian), or body weight (32 to 155 kg).
No clinically significant differences in the pharmacokinetics of brensocatib were observed based on mild, moderate, or severe renal impairment (eGFR 15-89 mL/min/1.73 m2); or mild, moderate, or severe hepatic impairment (Child-Pugh Class A, B, or C).
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
Strong CYP3A4 and P-gp Inhibitors: Brensocatib Cmax increased by 68% and AUC increased by 55% following concomitant administration with clarithromycin (a strong CYP3A4 and P-gp inhibitor) 500 mg twice daily for 6 days.
Moderate CYP3A4 and P-gp Inhibitors: Brensocatib Cmax increased by 53% and AUC increased by 32% following concomitant administration with verapamil (a moderate CYP3A4 and P-gp inhibitor) 240 mg once daily for 5 days.
Strong CYP3A4 Inducers: Brensocatib Cmax decreased by 15% and AUC decreased by 33% following concomitant administration with rifampin (a strong CYP3A4 inducer) 600 mg once daily for 9 days.
Acid-Reducing Agents: Brensocatib Cmax and AUC were unchanged following concomitant administration with esomeprazole (a proton-pump inhibitor) 40 mg once daily for 4 days.
CYP3A4 Substrates: No clinically significant differences in the pharmacokinetics of midazolam (a sensitive CYP3A4 substrate) are predicted when used concomitantly with brensocatib.
In vitro Studies
CYP450 Enzymes: Brensocatib is a substrate of CYP3A. Brensocatib does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, or CYP3A. Brensocatib is a weak CYP3A inducer, but not an inducer of CYP1A2, CYP2B6, CYP2C8, CYP2C9, or CYP2C19.
Transporter Systems: Brensocatib is a substrate of P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP), but is not a substrate of MATE1, MATE2-K, OAT1, OAT3, OATP1B1, OATP1B3, OCT1, and OCT2. Brensocatib is an inhibitor of BCRP, OATP1B, MATE1, and MATE2-K, but is not an inhibitor of P-gp, OAT1, OCT2, OAT3, or OATP1B3.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
A two-year study in Han Wistar rats and a 6-month study in Tg.rasH2 transgenic mice were conducted to assess the carcinogenic potential of brensocatib. No evidence of tumorigenicity was observed in male or female rats at an exposure approximately 56 times the MRHD on an AUC basis. No evidence of tumorigenicity was observed in male and female Tg.rasH2 mice at oral doses up to 50 mg/kg/day, the highest dose tested. Brensocatib was negative for genotoxicity in the following assays: in vitro bacterial reverse mutation (Ames) assay, in vitro mouse lymphoma mutation assay, and in vivo micronucleus assay in rats.
Oral administration of brensocatib to male and female rats at up to 50 and 100 mg/kg/day, respectively, had no adverse effects on fertility and reproductive performance indices (150 and 231 times the MRHD on an AUC basis, respectively).
14. Clinical Studies
The efficacy of BRINSUPRI was evaluated in two randomized, double-blind, placebo-controlled, parallel-group, multicenter, multinational clinical trials (ASPEN [NCT04594369] and WILLOW [NCT03218917]).
ASPEN was a 52-week trial that included 1721 adult and pediatric patients 12 years of age and older with NCFB (1680 adults and 41 pediatric patients 12 years of age to less than 18 years of age) who were randomized to BRINSUPRI 10 mg (n=583), BRINSUPRI 25 mg (n=575), or placebo (n=563) administered orally once daily.
WILLOW was a 24-week trial that included 256 adult patients with NCFB who were randomized to BRINSUPRI 10 mg (n=82), BRINSUPRI 25 mg (n=87), or placebo (n=87) administered orally once daily.
In both ASPEN and WILLOW, all adult patients had a history of confirmed NCFB by chest computed tomography with at least 2 documented pulmonary exacerbations (PEx) prior to screening in the past 12 months. In ASPEN, pediatric patients 12 years of age and older had at least one PEx in the prior 12 months.
Demographics and baseline characteristics of patients in ASPEN and WILLOW are provided in Table 2.
Table 2. Demographics and Baseline Characteristics of Patients in ASPEN and WILLOW:
| ASPEN (N=1721) | WILLOW (N=256) | |
|---|---|---|
| Age (years), mean (SD) | 60 (16) | 64 (12) |
| Female n (%) | 1107 (64) | 174 (68) |
| White n (%) | 1266 (74) | 225 (88) |
| Black or African American n (%) | 10 (1) | 4 (2) |
| Asian n (%) | 191 (11) | 23 (9) |
| Hispanic or Latino n (%) | 511 (30) | 6 (2) |
| ≥3 PEx in prior 12 months n (%) | 502 (29) | 84 (33) |
| Former smoker n (%) | 510 (30) | 86 (34) |
| ppFEV1 post-bronchodilator, mean (SD) | 74 (23) | 68 (24) |
| Sputum positive for Pseudomonas aeruginosa n (%) | 607 (35) | 89 (35) |
| Chronic macrolide therapy n (%) | 329 (19) | 40 (16) |
Abbreviations: N, number of patients in the intent-to-treat analysis set; n, number of patients; PEx, pulmonary exacerbations; pp, percent predicted; FEV1, forced expiratory volume in 1 second; SD, standard deviation
ASPEN
The primary efficacy endpoint in ASPEN was the annualized rate of PEx over the 52-week treatment period.
Pulmonary exacerbations were defined as worsening of 3 or more of the following major symptoms over 48 hours: increased cough, increased sputum volume or change in sputum consistency, increased sputum purulence, increased breathlessness, decreased exercise tolerance, fatigue and/or malaise, and hemoptysis, resulting in a healthcare provider's decision to prescribe systemic antibiotics. Pulmonary exacerbations were considered severe if requiring treatment with intravenous antibacterial drugs and/or resulted in hospitalization.
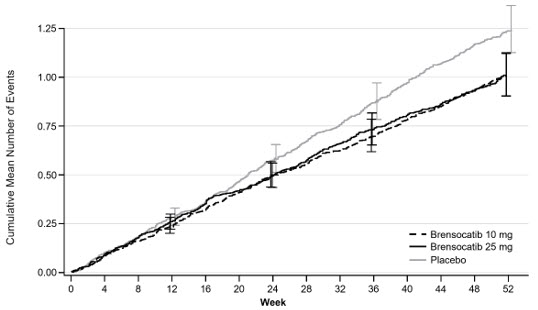
Treatment with BRINSUPRI 10 mg or 25 mg in patients with NCFB demonstrated reductions in the mean rate of PEx over 52 weeks compared with placebo (Figure 1). Table 3 provides the results of the annualized rate of PEx, and the results of the key secondary endpoints in ASPEN of time to first PEx, proportion of patients remaining exacerbation free throughout the 52-week treatment period, annualized rate of severe PEx, and the change from baseline in post-bronchodilator FEV1.
Table 3. Primary and Key Secondary Efficacy Analyses of Pulmonary Exacerbations and FEV1 Over 52 Weeks in ASPEN:
| Placebo (N=563) | BRINSUPRI 10 mg (N=583) | BRINSUPRI 25 mg (N=575) | |
|---|---|---|---|
| Annualized Rate of PEx | 1.29 | 1.02 | 1.04 |
| Rate Ratio (95% CI) | -- | 0.79 (0.68, 0.92) | 0.81 (0.69, 0.94) |
| Median Time to First PEx (weeks) | 36.71 | 49.00 | 50.71 |
| Hazard Ratio (95% CI) | -- | 0.81 (0.70, 0.95) | 0.83 (0.70, 0.97) |
| Proportion of Patients that were PEx Free at Week 52 (%) | 40.3 | 48.5 | 48.5 |
| Odds Ratio (95% CI) | -- | 1.41 (1.11, 1.81) | 1.40 (1.10, 1.79) |
| Annualized Rate of Severe PEx | 0.19 | 0.14 | 0.14 |
| Rate Ratio (95% CI) | -- | 0.74 (0.51, 1.09) | 0.74 (0.52, 1.06) |
| LS Mean Change from Baseline in Post- Bronchodilator FEV1 (mL) at Week 52 | -62 | -50 | -24 |
| Difference vs Placebo (95% CI) | -- | 11 (-14, 37) | 38 (11, 65) |
Abbreviations: FEV1, forced expiratory volume in 1 second; LS, least squares; PEx, pulmonary exacerbation
Figure 1. Cumulative Mean Number of Pulmonary Exacerbations through Week 52:
WILLOW
The primary efficacy endpoint in WILLOW was the time to first PEx over the 24-week treatment period. The time to first PEx was longer for patients receiving BRINSUPRI 10 mg and 25 mg compared to placebo (hazard ratio BRINSUPRI 10 mg and 25 mg versus placebo; 0.58 and 0.62, respectively; 95% CI: 0.35 to 0.95 and 0.38 to 0.99, respectively).
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.