CAMPTO Concentrate for solution for infusion Ref.[8066] Active ingredients: Irinotecan
Source: Medicines & Healthcare Products Regulatory Agency (GB) Revision Year: 2018 Publisher: Pfizer Limited, Ramsgate Road, Sandwich, Kent, CT13 9NJ, UK
Pharmacodynamic properties
Pharmacotherapeutic group: Cytostatic topoisomerase I inhibitor
ATC Code: L01XX19
Mechanism of action
Experimental data
Irinotecan is a semi-synthetic derivative of camptothecin. It is an antineoplastic agent which acts as a specific inhibitor of DNA topoisomerase I. It is metabolised by carboxylesterase in most tissues to SN-38, which was found to be more active than irinotecan in purified topoisomerase I and more cytotoxic than irinotecan against several murine and human tumour cell lines. The inhibition of DNA topoisomerase I by irinotecan or SN-38 induces single-strand DNA lesions which blocks the DNA replication fork and are responsible for the cytotoxicity. This cytotoxic activity was found time-dependent and was specific to the S phase.
In vitro, irinotecan and SN-38 were not found to be significantly recognised by the P-glycoprotein MDR, and displays cytotoxic activities against doxorubicin and vinblastine resistant cell lines.
Furthermore, irinotecan has a broad antitumor activity in vivo against murine tumour models (P03 pancreatic ductal adenocarcinoma, MA16/C mammary adenocarcinoma, C38 and C51 colon adenocarcinomas) and against human xenografts (Co-4 colon adenocarcinoma, Mx-1 mammary adenocarcinoma, ST-15 and SC-16 gastric adenocarcinomas). Irinotecan is also active against tumours expressing the P-glycoprotein MDR (vincristine- and doxorubicin-resistant P388 leukaemias).
Beside the antitumor activity of CAMPTO, the most relevant pharmacological effect of irinotecan is the inhibition of acetylcholinesterase.
Clinical data
In combination therapy for the first-line treatment of metastatic colorectal carcinoma
In combination therapy with Folinic Acid and 5-Fluorouracil
A phase III study was performed in 385 previously untreated metastatic colorectal cancer patients treated with either every 2 weeks schedule (see section 4.2) or weekly schedule regimens. In the every 2 weeks schedule, on day 1, the administration of CAMPTO at 180 mg/m² once every 2 weeks is followed by infusion with folinic acid (200 mg/m² over a 2-hour intravenous infusion) and 5-fluorouracil (400 mg/m² as an intravenous bolus, followed by 600 mg/m² over a 22-hour intravenous infusion). On day 2, folinic acid and 5-fluorouracil are administered at the same doses and schedules. In the weekly schedule, the administration of CAMPTO at 80 mg/m² is followed by infusion with folinic acid (500 mg/m² over a 2-hour intravenous infusion) and then by 5-fluorouracil (2300 mg/m² over a 24-hour intravenous infusion) over 6 weeks.
In the combination therapy trial with the 2 regimens described above, the efficacy of CAMPTO was evaluated in 198 treated patients:
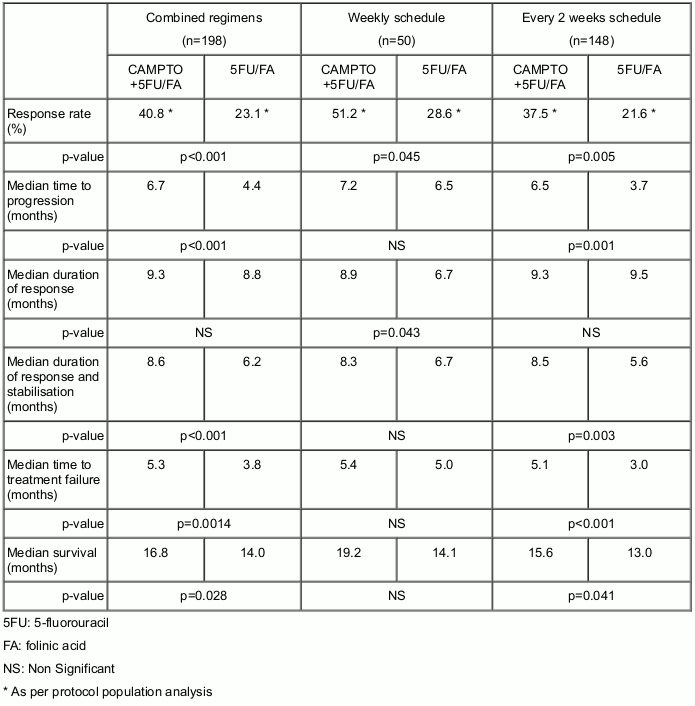
In the weekly schedule, the incidence of severe diarrhoea was 44.4% in patients treated by CAMPTO in combination with 5FU/FA and 25.6% in patients treated by 5FU/FA alone. The incidence of severe neutropenia (neutrophil count <500 cells/mm³) was 5.8% in patients treated by CAMPTO in combination with 5FU/FA and in 2.4% in patients treated by 5FU/FA alone.
Additionally, median time to definitive performance status deterioration was significantly longer in CAMPTO combination group than in 5FU/FA alone group (p=0.046).
Quality of life was assessed in this phase III study using the EORTC QLQ-C30 questionnaire. Time to definitive deterioration constantly occurred later in the CAMPTO groups. The evolution of the Global Health Status/Quality of life was slightly better in CAMPTO combination group although not significant, showing that efficacy of CAMPTO in combination could be reached without affecting the quality of life.
In combination therapy with bevacizumab
A phase III randomised, double-blind, active-controlled clinical trial evaluated bevacizumab in combination with CAMPTO/5FU/FA as first-line treatment for metastatic carcinoma of the colon or rectum (Study AVF2107g). The addition of bevacizumab to the combination of CAMPTO/5FU/FA resulted in a statistically significant increase in overall survival. The clinical benefit, as measured by overall survival, was seen in all pre-specified patient subgroups, including those defined by age, sex, performance status, location of primary tumour, number of organs involved, and duration of metastatic disease. Refer also to the bevacizumab summary of product characteristics. The efficacy results of Study AVF2107g are summarised in the table below.
| AVF2107g | |||
|---|---|---|---|
| Arm 1 CAMPTO/5FU/FA + Placebo | Arm 2 CAMPTO/5FU/FA + Avastina | ||
| Number of Patients | 411 | 402 | |
| Overall survival | |||
| Median time (months) | 15.6 | 20.3 | |
| 95% Confidence Interval | 14.29–16.99 | 18.46–24.18 | |
| Hazard ratiob | 0.660 | ||
| p-value | 0.00004 | ||
| Progression-free survival | |||
| Median time (months) | 6.2 | 10.6 | |
| Hazard ratio | 0.54 | ||
| p-value | <0.0001 | ||
| Overall response rate | |||
| Rate (%) | 34.8 | 44.8 | |
| 95% CI | 30.2–39.6 | 39.9–49.8 | |
| p-value | 0.0036 | ||
| Duration of response | |||
| Median time (months) | 7.1 | 10.4 | |
| 25–75 percentile (months) | 4.7–11.8 | 6.7–15.0 | |
a 5mg/kg every 2 weeks.
b Relative to control arm.
In combination therapy with cetuximab
EMR 62 202-013: This randomised study in patients with metastatic colorectal cancer who had not received prior treatment for metastatic disease compared the combination of cetuximab and irinotecan plus infusional 5-fluorouracil/folinic acid (5-FU/FA) (599 patients) to the same chemotherapy alone (599 patients). The proportion of patients with KRAS wild-type tumours from the patient populations evaluable for KRAS status comprised 64%.
The efficacy data generated in this study are summarised in the table below:
| Overall population | KRAS wild-type population | |||
|---|---|---|---|---|
| Variable/statistic | Cetuximab plus FOLFIRI (N=599) | FOLFIRI (N=599) | Cetuximab plus FOLFIRI (N=172) | FOLFIRI (N=176) |
| ORR | ||||
| % (95%CI) | 46.9 (42.9, 51.0) | 38.7 (34.8, 42.8) | 59.3 (51.6, 66.7) | 43.2 (35.8, 50.9) |
| p-value | 0.0038 | 0.0025 | ||
| PFS | ||||
| Hazard Ratio (95% CI) | 0.85 (0.726, 0.998) | 0.68 (0.501, 0.934) | ||
| p-value | 0.0479 | 0.0167 | ||
CI = confidence interval; FOLFIRI = irinotecan plus infusional 5-FU/FA; ORR = objective response rate (patients with complete response or partial response); PFS = progression-free survival time
In combination therapy with capecitabine
Data from a randomised, controlled phase III study (CAIRO) support the use of capecitabine at a starting dose of 1,000 mg/m² for 2 weeks every 3 weeks in combination with irinotecan for the first-line treatment of patients with metastatic colorectal cancer. Eight hundred twenty (820) patients were randomised to receive either sequential treatment (n=410) or combination treatment (n=410). Sequential treatment consisted of first-line treatment with capecitabine (1,250 mg/m² twice daily for 14 days), second-line irinotecan (350 mg/m² on day 1), and third-line combination of capecitabine (1,000 mg/m² twice daily for 14 days) with oxaliplatin (130 mg/m² on day 1). Combination treatment consisted of first-line treatment of capecitabine (1,000 mg/m² twice daily for 14 days) combined with irinotecan (250 mg /m² on day 1) (XELIRI) and second-line capecitabine (1,000 mg/m² twice daily for 14 days) plus oxaliplatin (130 mg/m² on day 1). All treatment cycles were administered at intervals of 3 weeks. In first-line treatment the median progression-free survival in the intent-to-treat population was 5.8 months (95%CI, 5.1 -6.2 months) for capecitabine monotherapy and 7.8 months (95%CI, 7.0-8.3 months) for XELIRI (p=0.0002).
Data from an interim analysis of a multicentre, randomised, controlled phase II study (AIO KRK 0604) support the use of capecitabine at a starting dose of 800 mg/m² for 2 weeks every 3 weeks in combination with irinotecan and bevacizumab for the first-line treatment of patients with metastatic colorectal cancer. One hundred fifteen (115) patients were randomised to treatment with capecitabine combined with irinotecan (XELIRI) and bevacizumab: capecitabine (800 mg/m² twice daily for 2 weeks followed by a 7-day rest period), irinotecan (200 mg/m² as a 30 minute infusion on day 1 every 3 weeks), and bevacizumab (7.5 mg/kg as a 30 to 90 minute infusion on day 1 every 3 weeks); a total of 118 patients were randomised to treatment with capecitabine combined with oxaliplatin plus bevacizumab: capecitabine (1,000 mg/m² twice daily for two weeks followed by a 7-day rest period), oxaliplatin (130 mg/m² as a 2 hour infusion on day 1 every 3 weeks), and bevacizumab (7.5 mg/kg as a 30 to 90 minute infusion on day 1 every 3 weeks). Progression-free survival at 6 months in the intent-to-treat population was 80% (XELIRI plus bevacizumab) versus 74% (XELOX plus bevacizumab). Overall response rate (complete response plus partial response) was 45% (XELOX plus bevacizumab) versus 47% (XELIRI plus bevacizumab).
In monotherapy for the second-line treatment of metastatic colorectal carcinoma
Clinical phase II/III studies were performed in more than 980 patients in the every 3-week dosage schedule with metastatic colorectal cancer who failed a previous 5-FU regimen. The efficacy of CAMPTO was evaluated in 765 patients with documented progression on 5-FU at study entry.
| Phase III | ||||||
|---|---|---|---|---|---|---|
| CAMPTO versus supportive care | CAMPTO versus 5FU | |||||
| CAMPTO n=183 | Supportive care n=90 | p-values | CAMPTO n=127 | 5FU n=129 | p-values | |
| Progression-Free Survival at 6 months (%) | NA | NA | 33.5* | 26.7 | p=0.03 | |
| Survival at 12 months (%) | 36.2* | 13.8 | p=0.0001 | 44.8* | 32.4 | p=0.0351 |
| Median survival (months) | 9.2* | 6.5 | p=0.0001 | 10.8* | 8.5 | p=0.0351 |
NA=Non Applicable
* Statistically significant difference
In phase II studies, performed on 455 patients in the every 3-week dosage schedule, the progression- free survival at 6 months was 30 % and the median survival was 9 months. The median time to progression was 18 weeks.
Additionally, non-comparative phase II studies were performed in 304 patients treated with a weekly schedule regimen, at a dose of 125 mg/m² administered as an intravenous infusion over 90 minutes for 4 consecutive weeks followed by 2 weeks rest. In these studies, the median time to progression was 17 weeks and median survival was 10 months. A similar safety profile has been observed in the weekly-dosage schedule in 193 patients at the starting dose of 125 mg/m², compared to the every 3-week-dosage schedule. The median time of onset of the first liquid stool was on day 11.
In combination with cetuximab after failure of irinotecan-including cytotoxic therapy
The efficacy of the combination of cetuximab with irinotecan was investigated in two clinical studies. A total of 356 patients with EGFR-expressing metastatic colorectal cancer who had recently failed irinotecan-including cytotoxic therapy and who had a minimum Karnofsky performance status of 60, but the majority of whom had a Karnofsky performance status of ≥80 received the combination treatment.
EMR 62 202-007: This randomised study compared the combination of cetuximab and irinotecan (218 patients) with cetuximab monotherapy (111 patients).
IMCL CP02-9923: This single arm open-label study investigated the combination therapy in 138 patients.
The efficacy data from these studies are summarised in the table below:
| Study | N | ORR | DCR | PFS (months) | OS (months) | ||||
|---|---|---|---|---|---|---|---|---|---|
| n (%) | 95% CI | n (%) | 95% CI | Median | 95% CI | Median | 95% CI | ||
| Cetuximab + Irinotecan | |||||||||
| EMR 62 202-007 | 218 | 50 (22.9) | 17.5, 29.1 | 121 (55.5) | 48.6, 62.2 | 4.1 | 2.8, 4.3 | 8.6 | 7.6, 9.6 |
| IMCLCP02-9923 | 138 | 21 (15.2) | 9.7, 22.3 | 84 (60.9) | 52.2, 69.1 | 2.9 | 2.6, 4.1 | 8.4 | 7.2, 10.3 |
| Cetuximab | |||||||||
| EMR 62 202-007 | 111 | 12 (10.8) | 5.7, 18.1 | 36 (32.4) | 23.9, 42.0 | 1.5 | 1.4, 2.0 | 6.9 | 5.6, 9.1 |
CI= confidence interval; DCR= disease control rate (patients with complete response, partial response, or stable disease for at least 6 weeks); ORR= objective response rate (patients with complete response or partial response); OS= overall survival time; PFS= progression-free survival
The efficacy of the combination of cetuximab with irinotecan was superior to that of cetuximab monotherapy, in terms of objective response rate (ORR), disease control rate (DCR) and progression-free survival (PFS). In the randomised trial, no effects on overall survival were demonstrated (hazard ratio 0.91, p=0.48).
Patients with Reduced UGT1A1 Activity
Uridine diphosphate-glucuronosyl transferase 1A1 (UGT1A1) is involved in the metabolic deactivation of SN-38, the active metabolite of irinotecan to inactive SN-38 glucuronide (SN-38G). The UGT1A1 gene is highly polymorphic, resulting in variable metabolic capacities among individuals. One specific variation of the UGT1A1 gene includes a polymorphism in the promoter region known as the UGT1A1*28 variant. This variant and other congenital deficiencies in UGT1A1 expression (such as Crigler-Najjar and Gilbert's syndrome) are associated with reduced activity of this enzyme. Data from a meta-analysis indicate that individuals with Crigler-Najjar syndrome (types 1 and 2) or those who are homozygous for the UGT1A1*28 allele (Gilbert's syndrome) are at increased risk of haematological toxicity (Grades 3 and 4) following administration of irinotecan at moderate or high doses (>150 mg/m²). A relationship between UGT1A1 genotype and the occurrence of irinotecan induced diarrhoea was not established.
Patients known to be homozygous for UGT1A1*28 should be administered the normally indicated irinotecan starting dose. However, these patients should be monitored for haematologic toxicities. A reduced irinotecan starting dose should be considered for patients who have experienced prior haematologic toxicity with previous treatment. The exact reduction in starting dose in this patient population has not been established and any subsequent dose modifications should be based on a patient's tolerance of the treatment (see sections 4.2 and 4.4).
There is at present insufficient data to conclude on clinical utility of UGT1A1 genotyping.
Pharmacokinetic properties
Absorption
At the end of the infusion, at the recommended dose of 350 mg/m², the mean peak plasma concentrations of irinotecan and SN-38 were 7.7 µg/ml and 56 ng/ml, respectively, and the mean area under the curve (AUC) values were 34 µg.h/ml and 451 ng.h/ml, respectively. A large interindividual variability in pharmacokinetic parameters is generally observed for SN-38.
Distribution
The phase I study in 60 patients with a dosage regimen of a 30-minute intravenous infusion of 100 to 750 mg/m² every three weeks, the volume of distribution at steady state (Vss): 157 L/m².
In vitro, plasma protein binding for irinotecan and SN-38 was approximately 65% and 95%, respectively.
Biotransformation
Mass balance and metabolism studies with 14C-labelled drug have shown that more than 50% of an intravenously administered dose of irinotecan is excreted as unchanged drug, with 33% in the faeces mainly via the bile and 22% in urine.
Two metabolic pathways account each for at least 12% of the dose:
- Hydrolysis by carboxylesterase into active metabolite SN-38, SN-38 is mainly eliminated by glucuronidation, and further by biliary and renal excretion (less than 0.5% of the irinotecan dose) The SN-38 glucuronite is subsequently probably hydrolysed in the intestine.
- Cytochrome P450 3A enzymes-dependent oxidations resulting in opening of the outer piperidine ring with formation of APC (aminopentanoic acid derivate) and NPC (primary amine derivate) (see section 4.5).
Unchanged irinotecan is the major entity in plasma, followed by APC, SN-38 glucuronide and SN-38. Only SN-38 has significant cytotoxic activity.
Elimination
In a phase I study in 60 patients with a dosage regimen of a 30-minute intravenous infusion of 100 to 750 mg/m² every three weeks, irinotecan showed a biphasic or triphasic elimination profile. The mean plasma clearance was 15 L/h/m². The mean plasma half-life of the first phase of the triphasic model was 12 minutes, of the second phase 2.5 hours, and the terminal phase half-life was 14.2 hours. SN-38 showed a biphasic elimination profile with a mean terminal elimination half-life of 13.8 hours.
Irinotecan clearance is decreased by about 40% in patients with bilirubinemia between 1.5 and 3 times the upper normal limit. In these patients a 200 mg/m² irinotecan dose leads to plasma drug exposure comparable to that observed at 350 mg/m² in cancer patients with normal liver parameters.
Linearity/non-linearity
A population pharmacokinetic analysis of irinotecan has been performed in 148 patients with metastatic colorectal cancer, treated with various schedules and at different doses in phase II trials. Pharmacokinetic parameters estimated with a three compartment model were similar to those observed in phase I studies. All studies have shown that irinotecan (CPT-11) and SN-38 exposure increase proportionally with CPT-11 administered dose; their pharmacokinetics are independent of the number of previous cycles and of the administration schedule.
Pharmacokinetic/Pharmacodynamic relationship(s)
The intensity of the major toxicities encountered with CAMPTO (e.g. leukoneutropenia and diarrhoea) are related to the exposure (AUC) to parent drug and metabolite SN-38. Significant correlations were observed between haematological toxicity (decrease in white blood cells and neutrophils at nadir) or diarrhoea intensity and both irinotecan and metabolite SN-38 AUC values in monotherapy.
Preclinical safety data
Irinotecan and SN-38 have been shown to be mutagenic in vitro in the chromosomal aberration test on CHO-cells as well as in the in vivo micronucleus test in mice.
However, they have been shown to be devoid of any mutagenic potential in the Ames test.
In rats treated once a week during 13 weeks at the maximum dose of 150 mg/m² (which is less than half the human recommended dose), no treatment related tumours were reported 91 weeks after the end of treatment.
Single- and repeated-dose toxicity studies with CAMPTO have been carried out in mice, rats and dogs. The main toxic effects were seen in the haematopoietic and lymphatic systems. In dogs, delayed diarrhoea associated with atrophy and focal necrosis of the intestinal mucosa was reported. Alopecia was also observed in the dog.
The severity of these effects was dose-related and reversible.
Reproduction
Irinotecan was teratogenic in rats and rabbits at doses below the human therapeutic dose. In rats, pups born to treated animals with external abnormalities showed a decrease in fertility. This was not seen in morphologically normal pups. In pregnant rats there was a decrease in placental weight and in the offspring a decrease in fetal viability and increase in behavioural abnormalities.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.