CAPRELSA Film-coated tablet Ref.[8958] Active ingredients: Vandetanib
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: Sanofi B.V., Paasheuvelweg 25, 1105 BP Amsterdam, The Netherlands
Pharmacodynamic properties
Pharmacotherapeutic group: antineoplastic agent, protein kinase inhibitor
ATC Code: L01EX04
Mechanism of action and pharmacodynamic effects
Vandetanib is a potent inhibitor of vascular endothelial growth factor receptor-2 (VEGFR-2 also known as kinase insert domain containing receptor [KDR]), epidermal growth factor receptor (EGFR) and RET tyrosine kinases. Vandetanib is also a sub-micromolar inhibitor of vascular endothelial receptor-3 tyrosine kinase.
Vandetanib inhibits VEGF-stimulated endothelial cell migration, proliferation, survival and new blood vessel formation in in vitro models of angiogenesis. In addition, vandetanib inhibits epidermal growth factor (EGF)-stimulated EGF receptor tyrosine kinase in tumour cells and endothelial cells. Vandetanib inhibits EGFR-dependent cell proliferation and cell survival in vitro. Vandetanib also inhibits both wild type and the majority of mutated, activated forms of RET, and significantly inhibits the proliferation of MTC cell lines in vitro.
In vivo vandetanib administration reduced tumour cell-induced angiogenesis, tumour vessel permeability, tumour microvessel density, and inhibited tumour growth of a range of human xenograft tumour models in athymic mice. Vandetanib also inhibited the growth of MTC xenograft tumours in vivo.
The precise mechanism of action of vandetanib in locally advanced or metastatic MTC is unknown.
Clinical efficacy in adults
Clinical data from MTC
A randomised, double-blind, placebo-controlled study (Study 58) was conducted to demonstrate safety and efficacy of vandetanib 300 mg versus placebo. This study included 331 patients with unresectable locally advanced or metastatic MTC. Only patients with CTN ≥500 pg/mL (conventional units) or ≥146.3 pmol/L (international standard units) were enrolled. Of the patients enrolled in the study 10 patients on vandetanib and 4 on placebo (4% of all patients) had a World Health Organization performance status (WHO PS) score of ≥2 and 28 (12.1%) patients on vandetanib and 10 (10.1%) on placebo had cardiac impairment. Cardiac impairment was defined as patients with previous cardiovascular abnormality.
The primary objective of this study was to demonstrate an improvement in progression-free survival (PFS) with vandetanib compared to placebo. The secondary endpoints were evaluation of overall objective response rate (ORR), disease control rate (DCR) defined as, partial response (PR) or complete response (CR) or stable disease (SD) lasting at least 24 weeks, duration of response (DOR), time to worsening of pain based on Brief Pain Inventory (BPI) worst pain scale, and overall survival (OS). The PFS primary endpoint, ORR and DCR were based on centralized, independent blinded review of the imaging data. Biochemical response with vandetanib as compared to placebo as measured by CTN and CEA was also assessed as secondary endpoints.
Patients were treated with vandetanib or placebo until they reached objective disease progression. Upon objective disease progression based on the investigator's assessment, patients were discontinued from blinded study treatment and given the option to receive open-label vandetanib. Twenty-eight of the 231 patients (12.1%) on vandetanib and 3 of the 99 (3.0%) on placebo discontinued treatment because of an adverse event. Fourteen of the 28 patients (50%) who stopped vandetanib for an adverse event discontinued without a dose reduction. Five out of 6 patients (83%) with moderate renal failure who were treated with vandetanib had a dose reduction to 200 mg for adverse reaction; 1 patient required a further reduction to 100 mg.
The result of the primary analysis of PFS showed a statistically significant improvement in PFS for patients randomised to vandetanib compared to placebo (Hazard Ratio (HR) = 0.46; 95% Confidence Interval (CI) = 0.31-0.69; p=0.0001).
The median PFS for patients randomised to vandetanib has not been reached; however, based on statistical modelling of data observed up to the 43 rd percentile, the median PFS is predicted to be 30.5 months with 95% confidence interval 25.5 to 36.5 months. The median PFS for patients randomised to placebo was 19.3 months. At 12 months, the proportion of patients alive and progression-free was 192 (83%) for patients randomised to vandetanib and 63 (63%) for patients randomised to placebo. In the vandetanib arm, a total of 73 (32%) patients progressed: 64 (28%) by response evaluation criteria in solid tumours (RECIST) progression and 9 (4%) by death in the absence of progression. The remaining 158 patients (68%) were censored in the analysis of PFS. In the placebo arm, a total of 51 (51%) of patients had progressed: 46 (46%) by RECIST progression and 5 (5%) by death in the absence of progression. The remaining 49 patients (49%) were censored in the analysis of PFS.
Figure 1. Kaplan Meier plot of PFS:
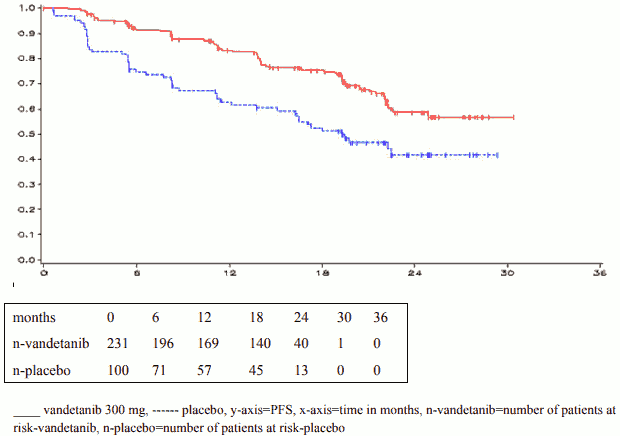
| months | 0 | 6 | 12 | 18 | 24 | 30 | 36 |
| n-vandetanib | 231 | 196 | 169 | 140 | 40 | 1 | 0 |
| n-placebo | 100 | 71 | 57 | 45 | 13 | 0 | 0 |
____ vandetanib 300 mg, ------ placebo, y-axis=PFS, x-axis=time in months, n-vandetanib=number of patients at risk-vandetanib, n-placebo=number of patients at risk-placebo
HR = 0.46, 95%CI (0.31-0.69), p = 0.0001
| PFS | N | Median PFS | HR | 95% CI | p-value |
| Vandetanib 300 mg | 73/231 (32%) | Not reached (predicted 30.5 months) | 0.46 | 0.31, 0.69 | 0.0001 |
| Placebo | 51/100 (51%) | 19.3 months |
Survival status and the median final overall survival (81.6 months in the vandetanib arm and 80.4 months in the placebo arm) were similar across both treatment arms. There was no statistically significant difference in final OS (HR 0.99, 95.002% CI 0.72, 1.38, p=0.9750). Results should be interpreted with caution due to the high percentage of patients in the placebo arm switching to open-label vandetanib (79.0% [79/100] of patients).
Most (95% of the patients) had metastatic disease. Fourteen patients treated with vandetanib, and 3 with placebo had unresectable locally advanced disease only. There is limited clinical experience with vandetanib in patients with unresectable locally advanced disease and without metastasis.
Statistically significant advantages were seen for vandetanib for the secondary endpoints of response rate, disease control rate, and biochemical response.
Table 3. Summary of other efficacy findings in study 58:
| ORRa | N | Response rate | ORb | 95% CI | p-value |
| Vandetanib 300 mg | 104/231 | 45% | 5.48 | 2.99, 10.79 | <0.0001 |
| Placebo | 13/100 | 13% | |||
| DCRa | N | Response rate | ORb | 95% CI | p-value |
| Vandetanib 300 mg | 200/231 | 87% | 2.64 | 1.48, 4.69 | 0.001 |
| Placebo | 71/100 | 71% | |||
| CTN Response | N | Response rate | ORb | 95% CI | p-value |
| Vandetanib 300 mg | 160/231 | 69% | 72.9 | 26.2, 303.2 | <0.0001 |
| Placebo | 3/100 | 3% | |||
| CEA Response< | N | Response rate | ORb | 95% CI | p-value |
| Vandetanib 300 mg | 119/231 | 52% | 52.0 | 16.0, 320.3 | <0.0001 |
| Placebo | 2/100 | 2% | |||
| OVERALL SURVIVAL | N | Median OS | HRc | 95% CI | p-value |
| Vandetanib 300 mg | 116/231 | 81.6 months | 0.99 | 0.72, 1.38 | 0.9750 |
| Placebo | 52/100 | 80.4 months |
a Overall response rate = complete + partial responses. Disease control rate = response rate + stable disease at 24 weeks. Intent-to-treat (ITT) analysis includes patients who received open-label vandetanib before progression according to the central read.
b OR = Odds Ratio. A value >1 favors vandetanib. The analysis was performed using a logistic regression model with treatment as the only factor.
c HR = Hazard Ratio. A value <1 favors vandetanib. The analysis was performed using a log rank test with treatment as the only factor.
N = Number of events/number of randomised patients
A statistically significant advantage was seen for vandetanib for the secondary endpoint of time to worsening of pain (derived as a composite endpoint using the worst pain score from BPI and patient reported opioid analgesic use) (vandetanib 49%, placebo 57%, HR 0.61, 97.5%CI 0.43-0.87, p<0.006: 8 vs. 3 months). There were no statistically significant differences observed for the exploratory endpoint of diarrhoea (reported as stool frequency).
RET mutation status
RET mutation status reanalysis in Study 58
In Study 58, RET mutation testing was initially performed by using the polymerase chain reaction (PCR) based Amplification Refractory Mutation System (ARMS) assay for the M918T mutation, and direct sequencing of DNA for mutations in exons 10, 11, 13, 14, 15 and 16 (site of M918T mutation) on all sporadic patients where DNA was available (297/298). For reanalysis of samples lacking M918T mutation, the RET sequences were enriched using a custom Agilent SureSelect reagent and sequenced on an Illumina sequencer. Data processing and automated calling of RET variants were carried out using the Broad Genome Analysis ToolKit (GATK) pipeline with manual curation of any difficult cases using Broad Integrative Genomics Viewer (IGV).
Initially, 79 patients had no M918T mutation identified. Of these 79 patients, 69 had enough tissue sample to allow a post-hoc reanalysis of RET mutation status based on new available assays. Most patients were reclassified as RET mutant (52/69) and 17/69 patients had no RET mutation (M918T or other) detected (11 with vandetanib and 6 with a placebo). Patients reclassified as RET mutant (N=52) were pooled with those 187 patients initially identified as RET mutant, leading to a total number of 239 RET mutant patients (172 treated with vandetanib and 67 treated with a placebo). Results were based on a blinded central review of imaging.
Table 4. Efficacy end-points in RET mutant patients:
| Efficacy end-point (Vandetanib vs placebo) | Patients with RET mutation (n=239) |
| Objective response rate | 51.7% vs 14.9% |
| Efficacy endpoint PFS HR (95% confidence interval) | 0.46 (0.29, 0.74) |
| 2-year PFS rate | 55.7% vs 40.1% |
Clinical efficacy in paediatric patients
A Phase I/II single-center open-label, single-arm study (Study IRUSZACT0098) assessed the activity of vandetanib in 16 patients with unresectable locally advanced or metastatic hereditary MTC. Characteristics of the patients at study entry were the following: mean age 14.2 years (range 9-17 years), 50% female, 50% male, 93.8% White, 26.7% Hispanic and 6.3% were Black. Most patients (81.3%) had undergone partial or total thyroidectomy prior to study entry. Starting vandetanib dose was 100mg/m²/day for all patients except for one who started at 150mg/m²/day. After having well tolerated the first 1 or 2 cycles of therapy (1 cycle = 28 days), the remaining patients continued on 100 mg/m² of treatment. The primary efficacy outcome was ORR according to RECIST v 1.0. The objective response rate observed was 43.8%, all of which were partial responses. 31.3% of patients had stable disease for at least 8 weeks. Disease Control Rate including best response or Stable Disease >24 weeks was 75.0%. There is no experience with Caprelsa in patients 5-8 years of age in this study.
Pharmacokinetic properties
Absorption
Following oral administration of vandetanib absorption is slow with peak plasma concentrations typically achieved at a median of 6 hours, range 4-10 hours, after dosing. Vandetanib accumulates approximately 8-fold on multiple dosing with steady state achieved from approximately 2 months.
Distribution
Vandetanib binds to human serum albumin and alpha-1-acid-glycoprotein with in vitro protein binding being approximately 90%. In ex vivo plasma samples from colorectal cancer patients at steady state exposure after 300 mg once daily, the mean percentage protein binding was 93.7% (range 92.2 to 95.7%). The pharmacokinetics of vandetanib at the 300 mg dose in MTC patients are characterised by a volume of distribution of approximately 7450 l.
Biotransformation
Following oral dosing of 14C-vandetanib, unchanged vandetanib and metabolites vandetanib N-oxide and N-desmethyl vandetanib were detected in plasma, urine and feces. A glucuronide conjugate was seen as a minor metabolite in excreta only. N-desmethyl-vandetanib is primarily produced by CYP3A4, and vandetanib-N-oxide by flavin-containing monooxygenase enzymes (FM01 and FMO3). N-desmethyl-vandetanib and vandetanib-N-oxide circulate at concentrations of approximately 11% and 1.4% of those of vandetanib.
Elimination
The pharmacokinetics of vandetanib at the 300 mg dose in MTC patients are characterised by a clearance of approximately 13.2 l/h. and plasma half-life of approximately 19 days. Within a 21 day collection period after a single dose of 14C-vandetanib, approximately 69% was recovered with 44% in faeces and 25% in urine. Excretion of the dose was slow and further excretion beyond 21 days would be expected based on the plasma half-life.
Special populations
Renal impairment
A single dose pharmacokinetic study in volunteers indicated that exposure to vandetanib is enhanced (up to 1.5, 1.6 and 2-fold) in mild, moderate and severe renal impaired subjects respectively compared to subjects with normal renal function (see sections 4.2, 4.4 and 4.5).
Hepatic impairment
A single dose pharmacokinetic study in volunteers indicated that hepatic impairment did not affect exposure to vandetanib. There is limited data in patients with hepatic impairment (serum bilirubin greater than 1.5 times upper limit of normal (see sections 4.2 and 4.4).
Food effect
Exposure to vandetanib is not affected by food.
Pharmacokinetics in paediatric population
The pharmacokinetic parameters of vandetanib in paediatrics MTC patients aged 9-17 years were similar to those in adults. Vandetanib exposure in children between 5-8 years old with glioma-related indications was comparable to MTC patients aged 9-18 years. Dosing at 100mg/m 2/day of the indicated posology (function of BSA) in paediatrics delivers similar exposure to that achieved in adults at 300 mg daily.
Preclinical safety data
Vandetanib has shown no mutagenic or clastogenic potential.
In repeat-dose toxicity studies of up to 9 months duration, effects included emesis, body weight loss and diarrhoea in dogs and physeal dysplasia in young dogs and rats with open growth plates. In rats, effects on teeth, kidney and skin were noted. These findings occurred at clinically-relevant plasma concentrations, were largely reversible within 4 weeks of cessation of dosing and were attributable to inhibition of vascular endothelial growth factor receptor (VEGFR) or EGFR.
Effects noted in other studies included inhibition of human ether-à-go-go related gene (hERG) current and prolongation of QTc interval in dogs. Elevation of systolic and diastolic blood pressure was observed in rats and dogs. In mice, vandetanib was shown to delay but not prevent wound healing. Vandetanib also showed evidence of phototoxic potential in an in vitro cytotoxicity assay. In an animal model of wound-healing, mice dosed with vandetanib had reduced skin-breaking strength compared with controls. This suggests that vandetanib slows but does not prevent wound healing. The appropriate interval between discontinuation of vandetanib and subsequent elective surgery required to avoid the risks of impaired wound healing has not been determined. In clinical studies, a small number of patients had surgery while receiving vandetanib and there were no reported wound healing complications.
Reproductive toxicology
Vandetanib had no effect on fertility in male rats. In a female fertility study, there was a trend towards increased oestrus cycle irregularity, a slight reduction in pregnancy incidence and increase in implantation loss. In a repeat-dose toxicity study in rats, there was a decrease in the number of corpora lutea in the ovaries of rats given vandetanib for 1 month.
In rats, embryofoetal toxicity was evident as foetal loss, delayed foetal development, heart vessel abnormalities and precocious ossification of some skull bones. In a rat pre- and post-natal development study, at doses producing maternal toxicity during gestation and/or lactation, vandetanib increased pre-birth loss and reduced post-natal pup growth. Vandetanib was excreted into milk in rat and found in plasma of pups following dosing to lactating rats.
Carcinogenicity
Vandetanib has shown no carcinogenic potential effect in a 6 month carcinogenicity study in rasH2 transgenic mice. A 2-year carcinogenicity study in rats was impaired by low survival in the high dose female group and limited exposure of the animals to vandetanib; however, no carcinogenic effects were observed in the remaining animals.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.