CONTROLOC Gastro-resistant tablet / Solution for injection Ref.[115943] Active ingredients: Pantoprazole
Source: Health Sciences Authority (SG) Revision Year: 2022 Publisher: Product Owner: Takeda GmbH, Konstanz, Germany
Pharmacodynamic properties
Controloc: contains Pantoprazole, a proton pump inhibitors which inhibits the gastric H K ATPase which is responsible for acid secretion in the parietal cells of the stomach. Pantoprazole is white to off-white powder with a molecular weight of 432.4. Pantoprazole is freely soluble in water, very slightly soluble in phosphate buffer at pH 7.4, and practically insoluble in n-hexane. Pantoprazole is a racemic mixture with a melting point of 138°C.
The chemical name for Pantoprazole is sodium-5-(difluoromethoxy)-2-[[(3,4-dimethoxy-2-pyridinyl)methyl] sulfinyl]-1 H-benzimidazole sesquihydrate and is represented by the following chemical structure:
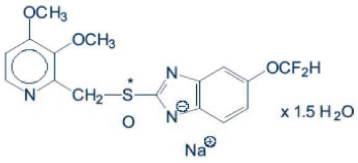
Empirical chemical structure: C16H14F2N3NaO4Sx1.5H2O
Pharmacotherapeutic / indication group / action mechanism
Selective proton pump inhibitor, substituted benzimidazole
(1) Mechanism of Action
Pantoprazole is a substituted benzimidazole which inhibits the secretion of hydrochloric acid in the stomach by specific action on the proton pumps of the parietal cells. Pantoprazole is converted to its active form in the acidic canaliculi in the parietal cells where it inhibits the H, K - ATPase enzyme, i.e. the final stage in the production of hydrochloric acid in the stomach.
The inhibition is dose-dependent and affects both basal and stimulated acid secretion. In most patients, freedom from symptoms is achieved in 2 weeks. As with other proton pump inhibitors and H2 receptor inhibitors, treatment with pantoprazole causes a reduced acidity in the stomach and thereby an increase in gastrin in proportion to the reduction in acidity. The increase in gastrin is reversible.
Since pantoprazole binds to the enzyme distal to the cell receptor level, the substance can affect hydrochloric acid secretion independently of stimulation by other substances (acetylcholine, histamine, gastrin). The effect is the same whether the active substance is given orally or intravenously. The fasting gastrin values increase under pantoprazole. On short-term use, in most cases they do not exceed the normal upper limit. During long-term treatment, gastrin levels double in most cases. An excessive increase, however, occurs only in isolated cases. As a result, a mild to moderate increase in the number of specific endocrine (ECL) cells in the stomach is observed in a minority of cases during long-term treatment (simple to adenomatoid hyperplasia). However, according to the studies conducted so far, the formation of carcinoid precursors (atypical hyperplasia) or gastric carcinoids as were found in animal experiments have not been observed in humans.
An influence of a long term treatment with pantoprazole exceeding one year cannot be completely ruled out on endocrine parameters of the thyroid and liver enzymes according to results in animal studies.
Pharmacokinetic properties
(1) Absorption
After ingestion, pantoprazole is rapidly absorbed into the bloodstream. On average the maximum serum concentrations (Cmax) of 1 to 1.5 μg/mL (pantoprazole 20 mg tablet) or 2 to 3 μg/mL (pantoprazole 40 mg tablet) are achieved at about 2 to 2.5 hours after administration. After single and repeated administration of pantoprazole, the pharmacokinetic characteristics of pantoprazole are very similar.
Both oral and I.V. administration of pantoprazole in the dose range of 10 mg to 80 mg result in linear serum pharmacokinetics. The absolute bioavailability from the tablet was found to be about 77%.
Concomitant intake of food had no relevant influence either on the AUC or on the Cmax and, thus, bioavailability. Only the variability of the lag-time will be increased by concomitant food intake.
(2) Distribution
Pantoprazole's serum protein binding is about 98%, and in keeping with this, pantoprazole has a low volume of distribution (about 0.15 l/kg) and limited tissue distribution.
(3) Metabolism
Pantoprazole is rapidly eliminated from the circulation and extensively metabolized in the liver. Metabolism occurs via oxidation by the CYP enzyme system, predominantly by CYP2C19 and CYP3A4 (Phase I metabolism, which is saturable). Pantoprazole undergoes further biotransformation by conjugation with sulphate, which involves the cytoplasmic enzyme sulphotransferase (phase II metabolism, which is not saturable), and which presents the main metabolism of pantoprazole.
(4) Excretion and elimination
About 80% of the metabolites of pantoprazole are eliminated via the renal route, the rest via the feces. None of the metabolites are considered as biologically active. The main metabolite in both the serum and urine is desmethylpantoprazole, which is conjugated with sulphate. T1/2 of the main metabolite is about 1.5 hour (which is not much longer than that of pantoprazole, 1 hour).
(5) Special populations
Impaired renal function
In patients with impaired renal function (including dialysis), pantoprazole showed no prolonged elimination half-life and no accumulation when compared with healthy subjects. No dose reduction is requested when pantoprazole is administered to patients with restricted kidney function (incl. dialysis patients). As with healthy subjects, pantoprazole's half-life is short. Only very small amounts of pantoprazole can be dialyzed. Although the main metabolite has a moderately delayed half-life (2 - 3h), excretion is still rapid and thus accumulation does not occur.
Impaired hepatic function
Although for patients with liver cirrhosis (classes A and B according to Child) the half-life values increased to between 3 and 6 h (pantoprazole 20 mg tablet) and the AUC values increased by a factor of 3 – 5 (pantoprazole 20 mg tablet), the maximum serum concentration only increased slightly by a factor of 1.3 (after oral administration) compared with healthy subjects.
A slight increase in AUC and Cmax in elderly volunteers compared with younger counterparts is also not clinically relevant.
Age, Gender, Race
As with other clinically used PPIs, a small percentage of the population (about 3% Caucasians, 20% Asians) shows slower elimination of pantoprazole (T1/2 being up to 10 hours as compared with 1hour). Such persons are known as poor metabolizers as a result of a deficiency of the CYP2C19 enzyme. In these individuals the metabolism of pantoprazole is probably mainly catalyzed by CYP3A4. After a single-dose administration of 40 mg pantoprazole, the mean area under the plasma concentration-time curve was approximately 6 times higher in poor metabolizers than in subjects having a functional CYP2C19 enzyme (extensive metabolizers). Mean peak plasma concentrations were increased by about 60%. These findings have no implications for the posology of pantoprazole.
Compared with younger subjects, slight increases in AUC and Cmax were noted after single and repeated oral administration of pantoprazole to healthy elderly subjects (age >65 years). However, no dose adjustment is generally necessary in elderly patients.
(6) Drug Interactions
Pantoprazole is metabolized in the liver via the CYP enzyme system. An interaction of pantoprazole with other drugs or compounds, which are metabolized using the same enzyme system, cannot be ruled out.
Nevertheless, in specific tests pantoprazole did not affect the clearance of several compounds metabolized by CYP enzymes. Vice-versa, all drugs that were tested regarding their potential influence on the pharmacokinetics of pantoprazole had no relevant effect.
No detectable interactions between pantoprazole and any other commonly prescribed co-medication tested so far were found.
Metabolism of pantoprazole occurs via oxidation by the CYP enzyme system, predominantly by CYP2C19 and CYP3A4. Interaction studies with drugs also metabolized by these pathways, like carbamazepine, diazepam, glibenclamide, nifedipine, phenytoin, and an oral contraceptive containing levonorgestrel and ethinyl estradiol did not reveal clinically significant interactions. Results from a range of interaction studies demonstrate that pantoprazole does not affect the metabolism of active substances metabolized by CYP1A2 (such as caffeine, theophylline), CYP2C9 (such as piroxicam, diclofenac, naproxen), CYP2D6 (such as metoprolol), or CYP2E1 (such as ethanol), or does not interfere with pglycoprotein related absorption of digoxin. There were no interactions with concomitantly administered antacids. Interaction studies have also been performed administering pantoprazole concomitantly with the respective antibiotics (clarithromycin, metronidazole, amoxicillin). No clinically relevant interactions were found.
(See Interaction with other medicinal products and other forms of interaction).
Preclinical safety data
Carcinogenesis, Mutagenesis, Impairment of Fertility
Non-clinical data reveal no special hazard to humans based on conventional studies of safety pharmacology, repeated dose toxicity and genotoxicity.
In the two-year carcinogenicity studies in rats, neuroendocrine neoplasms were found. In addition, squamous cell papillomas were found in the forestomach of rats. The mechanism leading to the formation of gastric carcinoids by substituted benzimidazoles has been carefully investigated and it can be concluded that it is a secondary reaction to the massively elevated serum gastrin levels occurring in the rat during chronic high dose treatment. In the two-year rodent studies, an increased number of liver tumors was observed in rats and in female mice and was interpreted as being due to pantoprazole's high metabolic rate in the liver.
Animal Toxicology and/or Pharmacology
A slight increase of neoplastic changes of the thyroid was observed in the group of rats receiving the highest dose (200 mg/kg). The occurrence of these neoplasms is associated with the pantoprazole-induced changes in the breakdown of thyroxine in the rat liver. As the therapeutic dose in man is low, no side effects to the thyroid glands are expected.
In animal reproduction studies, signs of fetotoxicity were observed at doses above 3 mg/kg.
Investigations revealed no evidence of impaired fertility or teratogenic effects. Crossing of the placenta was investigated in the rat and was found to increase with advanced gestation. As a result, the concentration of pantoprazole in the fetus is increased shortly before birth.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.