COPIKTRA Hard capsule Ref.[28036] Active ingredients: Duvelisib
Source: European Medicines Agency (EU) Revision Year: 2022 Publisher: Secura Bio Limited, 32 Molesworth Street, Dublin 2, Ireland
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: antineoplastic agents, phosphatidylinositol-3-kinase (Pi3K) inhibitors
ATC code: L01EM04
Mechanism of action
Duvelisib is a dual inhibitor of phosphatidylinositol 3-kinase p110δ (PI3K-δ) and PI3K-γ. PI3K-δ inhibition directly reduces proliferation and survival of malignant B-cell lines and primary CLL tumour cells, while PI3K-γ inhibition reduces the activity of CD4+ T cells and macrophages in the tumor microenvironment to support the malignant B cells. At 25 mg BID, the plasma levels of duvelisib may not be high enough to cause sustained inhibition of PI3K-γ, and the contribution of PI3K-γ inhibition to the efficacy may be limited.
Cardiac electrophysiology
The effect of multiple doses of duvelisib 25 and 75 mg BID on the corrected QT (QTc) interval was evaluated in patients with previously treated hematologic malignancies. Increases of >20 ms in the QTc interval were not observed.
Clinical efficacy in relapsed or refractory CLL/SLL
IPI-145-07
A randomised, multicenter, open-label trial (Study IPI-145-07) compared duvelisib versus ofatumumab in 319 adult patients with CLL (N=312) or SLL (N=7) after at least one prior therapy. Patients were not appropriate for treatment with a purine-based analogue regimen (per National Comprehensive Cancer Network or European Society for Medical Oncology guidelines), including relapse ≤36 month from a purine-based chemoimmunotherapy regimen or relapse ≤24 months from a purine-based monotherapy regimen. Patients who received prior BTK- or PI3K-inhibitors were excluded from the trial. None of the patients enrolled received prior BCL-2 inhibitor therapy. The study randomised patients with a 1:1 ratio to receive either duvelisib 25 mg BID until disease progression or unacceptable toxicity or ofatumumab for 7 cycles. Ofatumumab was administered intravenously at an initial dose of 300 mg, followed one week later by 2000 mg once weekly for 7 doses, and then 2000 mg once every 4 weeks for 4 additional doses. Treatment with ofatumumab beyond 7 cycles was not permitted, and no patients received more than 7 cycles of ofatumumab.
In the overall study population, (160 randomised to duvelisib, 159 to ofatumumab), the median patient age was 69 years (range: 39 to 90 years) with 68% of patients over 65 years, 60% were male, and 92% has an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1. 61% of patients had Rai stage of ≥ I, and 39% had Binet stage ≥ B. The percentage of patients with unmutated IGHV (Ig heavy chain V-111) was 71%. Thirty-eight percent (38%) received 1 prior line of therapy, and 62% received 2 or more prior lines. Ninety-four percent (94%) of patients received prior alkylator therapy, with 38% of patients receiving prior bendamustine therapy; 80% of patients received prior rituximab therapy. 60% in the duvelisib arm and 71% in the ofatumumab arm had prior purine analogue treatment (but were not refractory as defined by IwCLL). At baseline, 46% of patients had at least one tumour ≥5 cm, 24% of patients had a documented 17p deletion, 32% of patients had a documented 17p deletion and/or TP53 mutation, and 23% had a documented 11q deletion. The median time from initial diagnosis was 7 years (range: 0.3 to 34.7 years). The median time from most recent relapse/refractory diagnosis was 2.4 months (range: 0.2 to 80.2 months). The median time from most recent systemic therapy was 19.5 months (range: 0.5 to 148.8 months).
During randomised treatment, the median duration of exposure to duvelisib was 12 months (range: 0.2 to 37), with 72% of patients receiving at least 6 months and 49% receiving at least 12 months of duvelisib. The median duration of exposure to ofatumumab was 5 months (range: <0.1 to 6).
The approval of Copiktra is based on efficacy and safety analysis of patients with at least 2 prior lines of therapy, where the benefit:risk appeared greater in this more heavily pretreated population compared to the overall trial population.
In this subset of patients with at least 2 prior lines of therapy, (95 randomised to duvelisib, 101 to ofatumumab), the median patient age was 69 years (range: 40 to 90 years) with 70% of patients over 65 years, 59% were male, and 88% had an ECOG performance status of 0 or 1. 62% of the patients had Rai stage of ≥ I, and 38% had Binet stage ≥ B. The percentage of patients with unmutated IGHV (Ig heavy chain V-111) was 69%. Forty-six percent (46%) received 2 prior lines of therapy, and 54% received 3 or more prior lines. Ninety-six percent (96%) of patients received prior alkylator therapy, with 51% of patients receiving prior bendamustine therapy; 86% of patients received prior rituximab therapy. 70% in the duvelisib arm and 77% in the ofatumumab arm had prior purine analogue treatment (but were not refractory as defined by IwCLL). At baseline, 52% of patients had at least one tumour ≥5 cm, 22% of patients had a documented 17p deletion, 31% of patients had a documented 17p deletion and/or TP53 mutation, and 27% of patients had a documented 11q deletion. The median time from initial diagnosis was 8 years (range: 0.9 to 34.7 years). The median time from most recent relapse/refractory diagnosis was 2.6 months (range: 0.2 to 69 months). The median time from most recent systemic therapy was 15.5 months (range: 0.5 to 107.2 months).
During randomised treatment, the median duration of exposure to duvelisib was 13 months (range: 0.2 to 37), with 80% of patients receiving at least 6 months and 52% receiving at least 12 months of duvelisib. The median duration of exposure to ofatumumab was 5 months (range: <0.1 to 6).
Efficacy was based on the primary endpoint progression-free survival (PFS) as assessed by an Independent Review Committee (IRC). Patients on both arms were to continue to be followed for disease progression after discontinuation of randomized treatment until initiation of subsequent anticancer therapy. Other efficacy measures included overall response rate. The efficacy endpoints of overall response rate and overall survival were designated as key secondary efficacy endpoints and were to be tested sequentially only if the primary endpoint of PFS was significant.
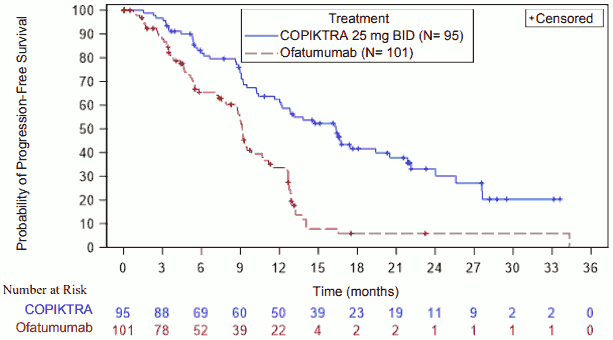
Results are presented in Table 3 and Figure 1 for the subset of patients with at least two prior therapies.
Table 3. Efficacy in CLL after at least two prior therapies (IPI-145-07):
| Outcome per IRC | Duvelisib N=95 | Ofatumumab N=101 |
|---|---|---|
| PFS | ||
| Median PFS (95% CI), monthsa | 16.4 (12.0, 20.5) | 9.1 (7.9, 10.7) |
| Hazard Ratio (95% CI),b Duvelisib/ofatumumab | 0.4 (0.27, 0.59) | |
| p-value | <0.0001 | |
| Response rate | ||
| ORR, n ()c (95 CI) | 75 (78.9) (70.7, 87.1) | 39 (38.6) (29.1, 48.1) |
| CR, n (%) | 0 | 0 |
| PR, n (%) | 75 (78.9) | 39 (38.6) |
| p-value | <0.0001 | |
| Overall Survival (OSd) | ||
| Median OS (95% CI), monthsa | 45.2 (35.9, 59.7) | 46.9 (33.3, 75.0) |
| Hazard Ratio (95% CI)b, Duvelisib/ofatumumab p-value | 1.1 (0.7, 1.6) 0.6065 | |
Abbreviations: CI = confidence interval; CR = complete response; IRC = Independent Review Committee; PFS = progression-free survival; PR = partial response
a Kaplan-Meier estimate
b Stratified Cox proportional hazards model using randomization strata as used for randomization
c IWCLL or revised IWG response criteria, with modification for treatment-related lymphocytosis
d OS analysis includes data from subjects who received ofatumumab on Study and subsequently received duvelisib in an extension study, based on intent-to-treat analysis. Subjects in both arms continued to be followed for OS after discontinuation of randomised treatment, regardless of subsequent therapies received. OS has been updated per the final analysis, with all subjects off study.
Table 4. Summary of PFS and response rates in subgroups therapy in patients with at least two prior therapies - (IPI-145-07):
| Outcome per IRC | Duvelisib | Ofatumumab |
|---|---|---|
| 17p deletion/TP53 mutation | N=29 | N=30 |
| Median PFS (95% CI), monthsa | 12.8 (8.9, 22.1) | 8.7 (5.3, 12.6) |
| Hazard Ratio (95% CI),b Duvelisib/ofatumumab | 0.36 (0.18, 0.72) | |
| ORR, (95% CI)c | 72.4 (56.1, 88.7) | 36.7 (19.4, 53.9) |
| Age ≥65 | N=68 | N=69 |
| Median PFS (95% CI), monthsa | 16.4 (10.4, 24.0) | 9.2 (8.7, 10.8) |
| Hazard Ratio (95% CI),b Duvelisib/ofatumumab | 0.38 (0.24, 0.61) | |
| ORR, (95% CI)c | 77.9 (68.1, 87.8) | 39.1 (27.6, 50.6) |
| Unmutated IGHV | N=65 | N=70 |
| Median PFS (95% CI), monthsa | 17.4 (12.0, 24.0) | 9.0 (7.3, 10.7) |
| Hazard Ratio (95% CI),b Duvelisib/ofatumumab | 0.27 (0.17, 0.45) | |
| ORR, (95% CI)c | 86.2 (77.8, 94.6) | 40 (28.5, 51.5) |
Abbreviations: CI = confidence interval; IRC = Independent Review Committee; PFS = progression-free survival
a Kaplan-Meier estimate
b Cox proportional hazards model
c IWCLL or revised IWG response criteria, with modification for treatment-related lymphocytosis
Figure 1. Kaplan-Meier curve of PFS per IRC in patients with at least two prior therapies (IPI-145-07):
Clinical efficacy in relapsed or refractory Follicular Lymphoma (FL)
IPI-145-06
Efficacy of duvelisib in patients with previously treated FL is based on a single-arm, multicenter trial (Study IPI-145-06). In this study, duvelisib 25 mg BID was administered in 129 patients with indolent B-cell non-Hodgkin lymphoma (iNHL, including: FL, n=83; SLL, n=28; and marginal zone lymphoma [MZL], n=18) who were refractory to rituximab and to either chemotherapy or radioimmunotherapy. Refractory disease was defined as less than a partial remission or relapse within 6 months after the last dose. The trial excluded patients with Grade 3b FL, large cell transformation, prior allogeneic transplant, and prior exposure to a PI3K inhibitor or to a Bruton’s tyrosine kinase inhibitor.
The median age was 65 years (range: 30 to 90 years) with 50% of subjects over 65 years and 14% of subjects age 75 or older, 68% were male, and 40% had bulky disease assessed at baseline (target lesion ≥5 cm). Patients had a median of 3 prior lines of therapy (range: 1 to 18), with 96% being refractory to their last therapy and 77% being refractory to 2 or more prior lines of therapy. Ninetyeight percent (98%) of patients were refractory to rituximab, and 91% were refractory to an alkylating agent. Most patients (approximately 75%) experienced early relapse (no response on treatment or progressive disease [PD] or time to next treatment less than 2 years) after their first treatment regimen. The median time from initial diagnosis was 4.5 years (range 4 months to 27 years). Most patients (95%) had an ECOG performance status of 0 or 1.
The median duration of exposure to duvelisib was 7 months (range: 0.4 to 45.5), with 53% of patients receiving at least 6 months and 26% receiving at least 12 months of duvelisib.
Efficacy was based on the primary endpoint of overall response rate. Secondary endpoints were progression-free survival, duration of response as assessed by an IRC and overall survival (Table 5).
Table 5. Efficacy in patients with at least two prior therapies, relapsed or refractory FL (IPI-145-06):
| Endpoint | |
|---|---|
| FL | N=73 |
| ORR, n (%)a | 29 (40) |
| 95% CI | (31, 54) |
| CR, n (%) | 0 |
| PR, n (%) | 29 (40) |
| Duration of response | |
| Range, months | 0.0+ to 41.9 |
| Median DOR (95% CI), monthsb | 10.01 (6.3, NE) |
Abbreviations: CI = confidence interval; CR = complete response; IRC = Independent Review Committee; ORR = overall response rate; PR = partial response
a Per IRC according to Revised International Working Group criteria
b Kaplan-Meier estimate
+ Denotes censored observation
Elderly
Clinical trials of duvelisib included 270 patients (61%) that were 65 years of age and older and 104 (24%) that were 75 years of age and older. No major differences in efficacy or safety were observed between patients less than 65 years of age and patients 65 years of age and older. No specific dose adjustment is required for elderly patients (aged ≥65 years) (see section 5.2).
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with duvelisib for the treatment of mature B cell malignancies for all subsets of the paediatric population from birth to less than 18 years of age (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Duvelisib exposure increased in a dose-proportional manner over a dose range of 8 mg to 75 mg (0.3 to 3 times the recommended dose) following a single dose. Dose proportionality was not estabilished after multiple doses.
At steady state following 25 mg BID administration of duvelisib in patients, the geometric mean (CV%) maximum concentration (Cmax) was 1.5 (64%) µg/mL and AUC was 7.9 (77%) µg•h/mL.
Absorption
The absolute bioavailability of 25 mg duvelisib after a single oral dose in healthy volunteers was 42%. The median time to peak concentration (Tmax) was observed at 1 to 2 hours in patients.
Effect of food
Duvelisib may be administered without regard to food. The administration of a single dose of duvelisib with a high-fat meal (fat accounted for approximately 50% of the total caloric content of the meal) decreased Cmax by approximately 37% and decreased the AUC by approximately 6%, relative to fasting conditions.
Distribution
Protein binding of duvelisib is greater than 95%. The mean blood-to-plasma ratio was 0.5. The geometric mean (CV%) apparent volume of distribution at steady state (Vss/F) is 28.5 L (62%).
Biotransformation
Duvelisib is primarily metabolized by cytochrome P450 CYP3A4. The major metabolite is IPI-656, which is pharmacologically inactive at clinically observed exposure levels.
Elimination
The geometric mean (CV%) apparent systemic clearance at steady-state is 4.2 L/hr (56%) in patients with lymphoma or leukaemia. The geometric mean (CV%) elimination half-life of duvelisib is 4.7 hours (57%) during 0-8 hours postdose.
Excretion
Following a single 25 mg oral dose of radiolabeled duvelisib, 79% of the radioactivity was excreted in feces (11% unchanged) and 14% was excreted in the urine (1% unchanged). These data have been determined in healthy subjects.
In vitro drug interaction studies
Duvelisib is a substrate of P-glycoprotein (P-gp) and breast cancer-resistant protein (BCRP). Duvelisib is highly absorbed following an oral dose and therefore no clinically relevant effect of P-gp and BCRP inhibitors is expected.
In vitro studies combined with human in vivo Pharmacokinetic (PK) data suggested that clinically relevant drug-drug interactions of duvelisib and its main metabolite IPI-656 with substrates of OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3, BCRP, or P-gp are unlikely. Therefore, interaction studies with Pgp, BCRP and CYP2C8 are considered not necessary.
Both duvelisib and IPI-656 were determined as direct inhibitors of CYP2C8 and CYP3A4 as well as metabolism-dependent inhibitors of CYP3A4 (Please refer to section 4.5). Simulations indicated that at supratherapeutic doses duvelisib can be a mild inhibitor of CYP2C8, which is considered unlikely to result in clinically relevant interactions.
Special populations
Age (18 to 90 years), sex, race, renal impairment (creatinine clearance 23 to 80 mL/min), hepatic impairment (Child Pugh Class A, B, and C) and body weight (40 to 154 kg) had no clinically significant effect on the exposure of duvelisib.
Duvelisib pharmacokinetics were highly variable in subjects with moderate and severe hepatic impairment.Geometric mean duvelisib AUC0-∞ in mild, moderate, and severely hepatically impaired subjects were lower (within 20%) compared to the exposure observed in healthy subjects and was 89%, 94%, and 81% of the exposure observed in healthy subjects and is not considered clinically significant. The exposures in moderately and severely impaired subjects were highly variable (CV% 46–67%) and these patients should be carefully monitored for adverse events (see section 4.4). Exposures obtained in cancer patients were approximately 2-fold higher than the exposures observed in healthy subjects.
5.3. Preclinical safety data
In repeat-dose toxicity studies in rat and cynomolgus monkey, adverse effects were mainly related to expected exaggarated pharmacology, including adverse effects on lymphoid tissues, bone marrow and haematology parameters at exposures of free duvelisib at 8 to 16 fold, corresponding to total duvelisib at 2 to 11 fold Maximum Recommended Human Dose (MRHD) of 25 mg BID in human.
Duvelisib did not cause genetic damage in in vitro or in vivo assays.
In dose range finding and pivotal embryo-fetal developmental toxicity studies in rat and rabbit, duvelisib (free fraction) induced embryo-fetal developmental toxicity only at free plasma exposures margins of >25 fold of 25 mg BID in human (MRHD), corresponding to 4 to 5 fold total plasma concentrations.
Fertility studies with duvelisib were not conducted. Histological findings in male and female rats were observed in the repeat dose toxicity studies and included testis (seminiferous epithelial atrophy, decreased weight, soft testes), and epididymis (small size, oligo/aspermia) in males and ovary (decreased weight) and uterus (atrophy) in females.
Carcinogenicity studies have not been conducted with duvelisib.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.