CRIXIVAN Hard capsule Ref.[8780] Active ingredients: Indinavir
Source: European Medicines Agency (EU) Revision Year: 2018 Publisher: Merck Sharp & Dohme B.V., Waarderweg 39, 2031 BN Haarlem, The Netherlands
Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in section 6.1.
Indinavir with or without ritonavir should not be administered concurrently with medicinal products with narrow therapeutic windows and which are substrates of CYP3A4. Inhibition of CYP3A4 by both CRIXIVAN and ritonavir could result in elevated plasma concentrations of these medicines, potentially causing serious or life-threatening reactions (see section 4.5).
CRIXIVAN with or without ritonavir should not be administered concurrently with amiodarone, terfenadine, cisapride, astemizole, quetiapine, alprazolam, triazolam, midazolam administered orally (for caution on parenterally administered midazolam, see section 4.5), pimozide, ergot derivatives, simvastatin or lovastatin (see section 4.4).
Combination of rifampicin with CRIXIVAN with or without concomitant low-dose ritonavir is contraindicated (see section 4.5). Concurrent use of indinavir with herbal preparations containing St John’s wort (Hypericum perforatum) is contraindicated (see section 4.5).
In addition, indinavir with ritonavir must not be administered with alfuzosin, meperidine, piroxicam, propoxyphene, bepridil, encainide, flecanide, propafenone, quinidine, fusidic acid, clozapine, clorazepate, diazepam, estazolam and flurazepam.
Indinavir must not be given with ritonavir to patients with decompensated liver disease as ritonavir is principally metabolized and eliminated by the liver (see section 4.4).
When CRIXIVAN is used with ritonavir, consult the Summary of Product Characteristics of ritonavir for additional contraindications.
Special warnings and precautions for use
Nephrolithiasis and tubulointerstitial nephritis
Nephrolithiasis has occurred with indinavir therapy in adult patients with a cumulative frequency of 12.4% (range across individual trials: 4.7% to 34.4%). The cumulative frequency of nephrolithiasis events increases with increasing exposure to CRIXIVAN; however, the risk over time remains relatively constant. In some cases, nephrolithiasis has been associated with renal insufficiency or acute renal failure; in the majority of these cases renal insufficiency and acute renal failure were reversible. If signs and symptoms of nephrolithiasis, including flank pain with or without haematuria (including microscopic haematuria) occur, temporary interruption of therapy (e.g. for 1-3 days) during the acute episode of nephrolithiasis or discontinuation of therapy may be considered. Evaluation may consist of urinalysis, serum BUN and creatinine, and ultrasound of the bladder and kidneys. Adequate hydration is recommended in all patients on indinavir (see sections 4.2 and 4.8).
Medical management in patients with one or more episodes of nephrolithiasis must include adequate hydration and may include temporary interruption of therapy (e.g., 1 to 3 days) during the acute episode of nephrolithiasis or discontinuation of therapy.
Cases of interstitial nephritis with medullary calcification and cortical atrophy have been observed in patients with asymptomatic severe leucocyturia (> 100 cells/high power field). In patients at increased risk, urinary screening should be considered. If persistent severe leucocyturia is found, further investigation might be warranted.
Medicinal product interactions
Indinavir should be used cautiously with other medicinal products that are potent inducers of CYP3A4. Co-administration may result in decreased plasma concentrations of indinavir and as a consequence an increased risk for suboptimal treatment and facilitation of development of resistance (see section 4.5).
If indinavir is given with ritonavir, the potential interaction may be increased. The Interactions section of the SPC for ritonavir should also be consulted for information about potential interactions.
Atazanavir as well as indinavir are associated with indirect (unconjugated) hyperbilirubinemia due to inhibition of UDP-glucuronosyltransferase (UGT). Combinations of atazanavir with or without ritonavir and Crixivan have not been studied and co-administration of these medicinal products is not recommended due to risk of worsening of these adverse reactions.
Concomitant use of indinavir with lovastatin or simvastatin is not recommended due to an increased risk of myopathy including rhabdomyolysis. Based on an interaction study with lopinavir/ritonavir, combination of rosuvastatin and protease inhibitors is not recommended. Caution must also be exercised if indinavir is used concurrently with atorvastatin. The interaction of indinavir or indinavir/ritonavir with pravastatin or fluvastatin is not known (see section 4.5).
Co-administration of CRIXIVAN with sildenafil, tadalafil and vardenafil (PDE5 inhibitors) are expected to substantially increase the plasma concentrations of these compounds and may result in an increase in PDE5 inhibitor-associated adverse events, including hypotension, visual changes, and priapism (see section 4.5).
HIV Transmission
While effective viral suppression with antiretroviral therapy has been proven to substantially reduce the risk of sexual transmission, a residual risk cannot be excluded. Precautions to prevent transmission should be taken in accordance with national guidelines.
Acute haemolytic anaemia
Acute haemolytic anaemia has been reported which in some cases was severe and progressed rapidly. Once a diagnosis is apparent, appropriate measures for the treatment of haemolytic anaemia should be instituted which may include discontinuation of indinavir.
Weight and metabolic parameters
An increase in weight and in levels of blood lipids and glucose may occur during antiretroviral therapy. Such changes may in part be linked to disease control and life style. For lipids, there is in some cases evidence for a treatment effect, while for weight gain there is no strong evidence relating this to any particular treatment. For monitoring of blood lipids and glucose reference is made to established HIV treatment guidelines. Lipid disorders should be managed as clinically appropriate.
Liver disease
The safety and efficacy of indinavir has not been established in patients with significant underlying liver disorders. Patients with chronic hepatitis B or C and treated with combination antiretroviral therapy are at an increased risk for severe and potentially fatal hepatic adverse reactions. In case of concomitant antiviral therapy for hepatitis B or C, please refer also to the relevant product information for these medicinal products.
Patients with pre-existing liver dysfunction including chronic active hepatitis have an increased frequency of liver function abnormalities during combination antiretroviral therapy and should be monitored according to standard practice. If there is evidence of worsening liver disease in such patients, interruption or discontinuation of treatment must be considered.
An increased incidence of nephrolithiasis has been observed in patients with underlying liver disorders when treated with indinavir.
Immune Reactivation Syndrome
In HIV-infected patients with severe immune deficiency at the time of institution of combination antiretroviral therapy (CART), an inflammatory reaction to asymptomatic or residual opportunistic pathogens may arise and cause serious clinical conditions, or aggravation of symptoms. Typically, such reactions have been observed within the first few weeks or months of initiation of CART. Relevant examples are cytomegalovirus retinitis, generalised and/or focal mycobacterial infections, and Pneumocystis carinii pneumonia. Any inflammatory symptoms should be evaluated and treatment instituted when necessary.
Autoimmune disorders (such as Graves' disease and autoimmune hepatitis) have also been reported to occur in the setting of immune reactivation; however, the reported time to onset is more variable and these events can occur many months after initiation of treatment.
Patients with coexisting conditions
There have been reports of increased bleeding, including spontaneous skin haematomas and haemarthroses, in haemophiliac patients type A and B treated with PIs. In some patients additional factor VIII was given. In more than a half of the reported cases, treatment with PIs was continued or re-introduced if treatment had been discontinued. A causal relationship has been evoked, although the mechanism of action has not been elucidated. Haemophiliac patients should therefore be made aware of the possibility of increased bleeding.
Patients with mild-to-moderate hepatic insufficiency due to cirrhosis will require a dose reduction of indinavir due to decreased metabolism of indinavir (see section 4.2). Patients with severe hepatic impairment have not been studied. In the absence of such studies, caution should be exercised as increased levels of indinavir may occur.
Safety in patients with impaired renal function has not been studied; however, less than 20% of indinavir is excreted in the urine as unchanged medicinal product or metabolites (see section 4.2).
Osteonecrosis
Although the etiology is considered to be multifactorial (including corticosteroid use, alcohol consumption, severe immunosuppression, higher body mass index), cases of osteonecrosis have been reported particularly in patients with advanced HIV-disease and/or long-term exposure to combination antiretroviral therapy (CART). Patients should be advised to seek medical advice if they experience joint aches and pain, joint stiffness or difficulty in movement.
Lactose
This medicinal product contains 299.2 mg of lactose in each 800 mg dose (maximum single dose). Patients with rare hereditary problems of galactose intolerance, the Lapp lactase deficiency or glucose-galactose malabsorption should not take this medicine.
Interaction with other medicinal products and other forms of interaction
The metabolism of indinavir is mediated by the cytochrome P450 enzyme CYP3A4. Therefore, other substances that either share this metabolic pathway or modify CYP3A4 activity may influence the pharmacokinetics of indinavir. Similarly, indinavir might also modify the pharmacokinetics of other substances that share this metabolic pathway. Boosted indinavir (indinavir with ritonavir) may have additive pharmacokinetic effects on substances that share the CYP3A4 pathway as both ritonavir and indinavir inhibit the cytochrome P450 enzyme CYP3A4.
Indinavir with or without ritonavir should not be administered concurrently with medicinal products with narrow therapeutic windows and which are substrates of CYP3A4. Inhibition of CYP3A4 by both CRIXIVAN and ritonavir could result in elevated plasma concentrations of these medicines, potentially causing serious or life-threatening reactions. CRIXIVAN with or without ritonavir should not be administered concurrently with amiodarone, terfenadine, cisapride, astemizole, quetiapine, alprazolam, triazolam, midazolam administered orally (for caution on parenterally administered midazolam, see Table 1 and 2 below), pimozide, ergot derivatives, simvastatin or lovastatin. In addition, indinavir with ritonavir should not be administered with alfuzosin, meperidine, piroxicam, propoxyphene, bepridil, encainide, flecanide, propafenone, quinidine, fusidic acid, clozapine, clorazepate, diazepam, estazolam and flurazepam.
Concurrent use of indinavir with rifampicin or herbal preparations containing St John’s wort (Hypericum perforatum) is contraindicated.
Medicinal products listed above are not repeated in Table 1 and 2 unless specific interaction data is available.
Refer also to sections 4.2 and 4.3.
Table 1. Interactions and dose recommendations with other medical products – UNBOOSTED INDINAVIR:
Interactions between indinavir and other medicinal products are listed in the tables below (increase is indicated as “↑”, decrease as “↓”, no change (≤ +/- 20%) as “↔”, single dose as “SD”, once daily as “QD”, twice daily as “BID”, three times daily as “TID”, and four times daily as “QID”).

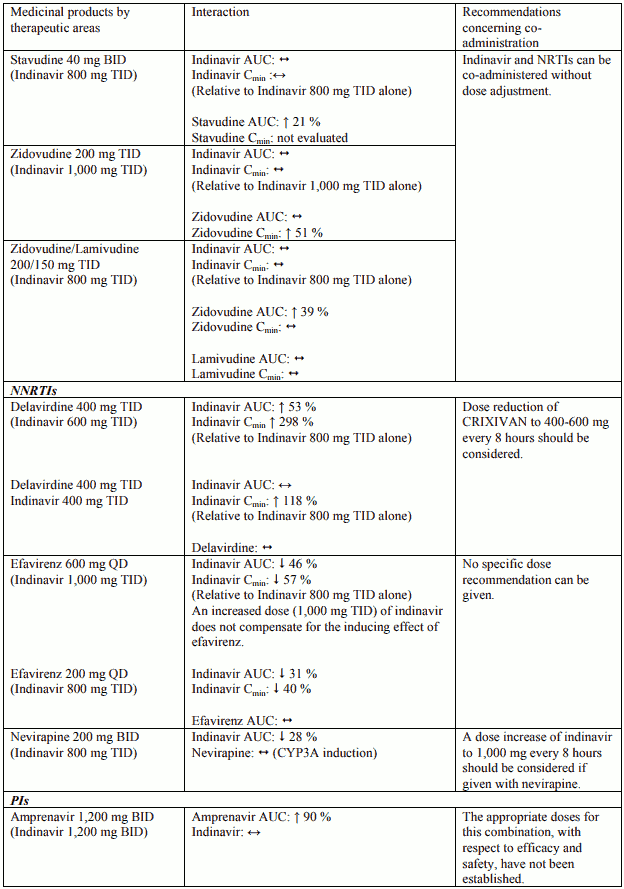

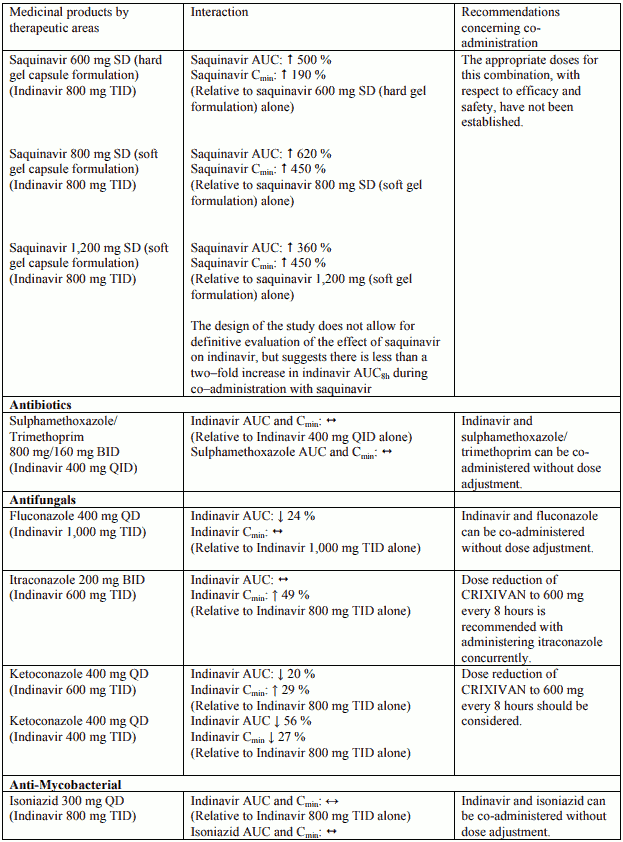
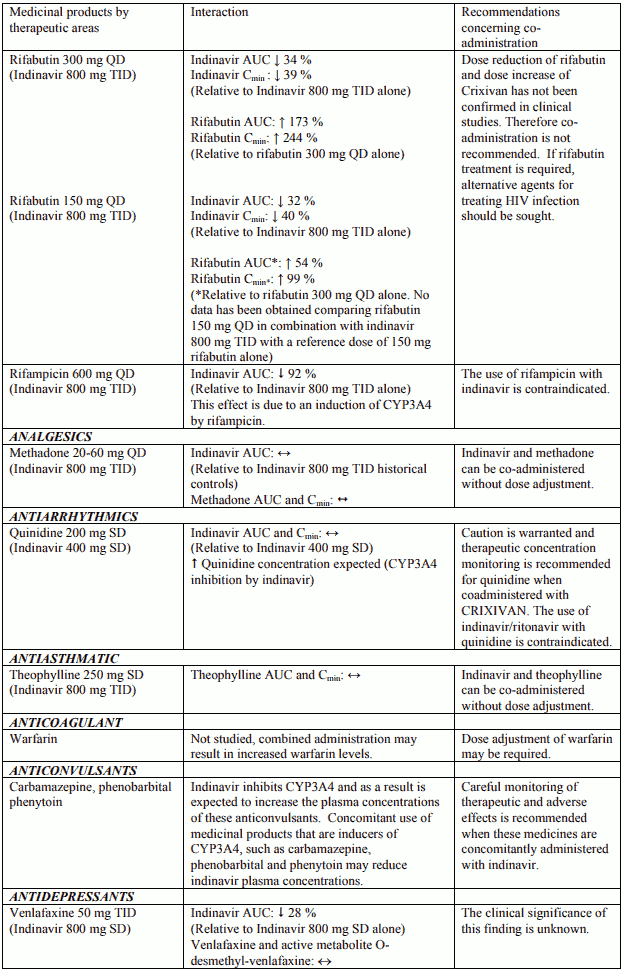
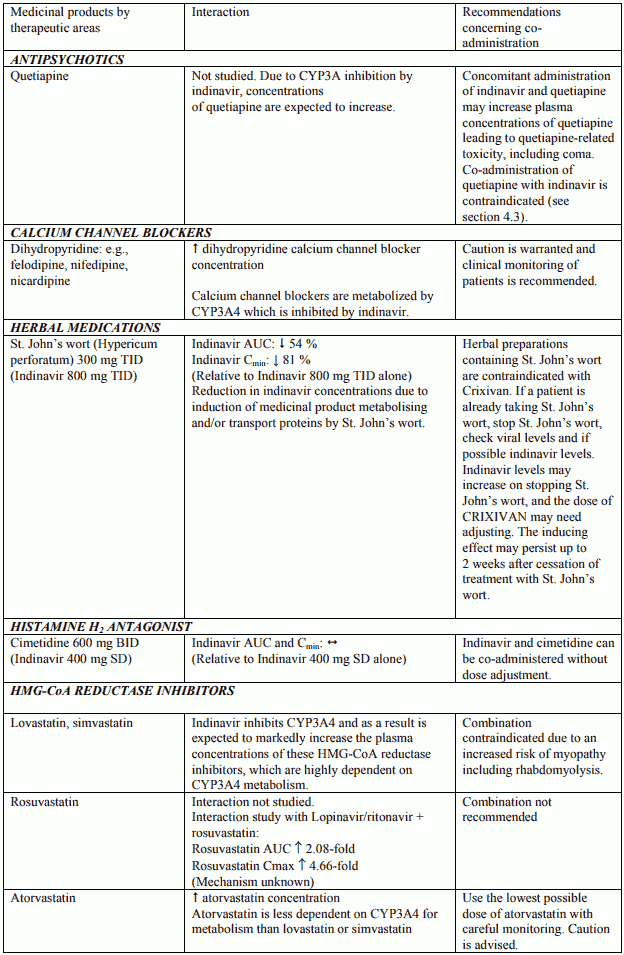
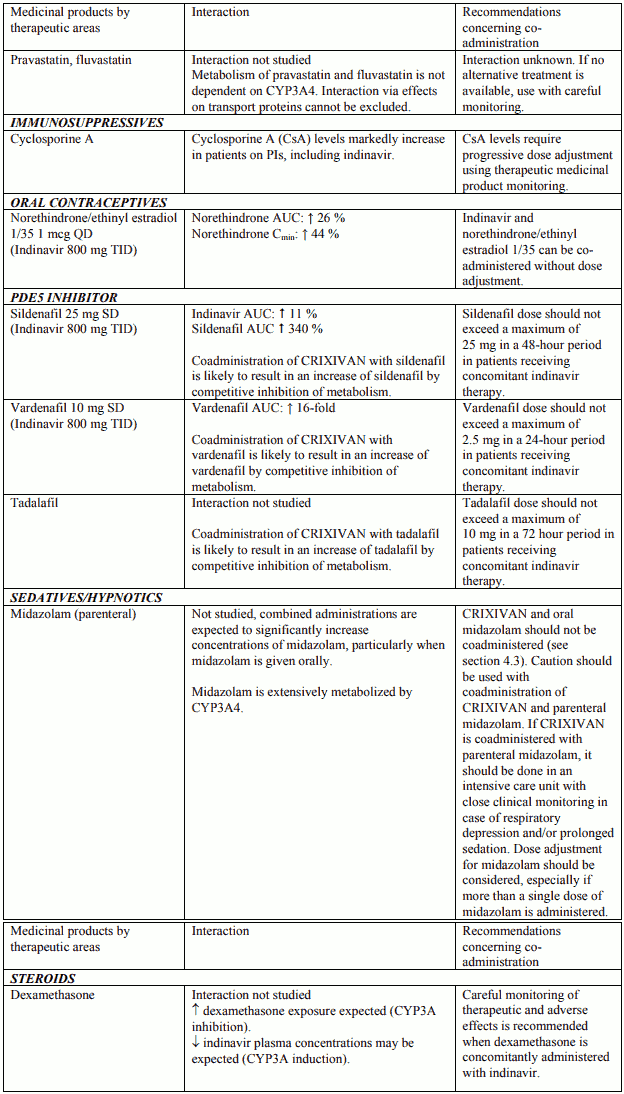
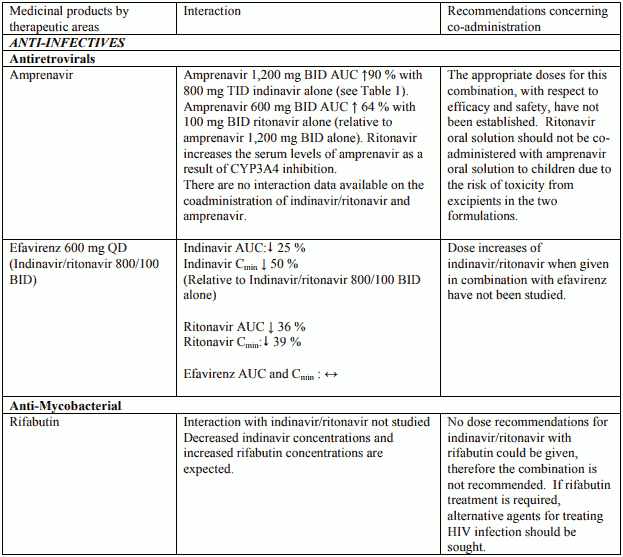
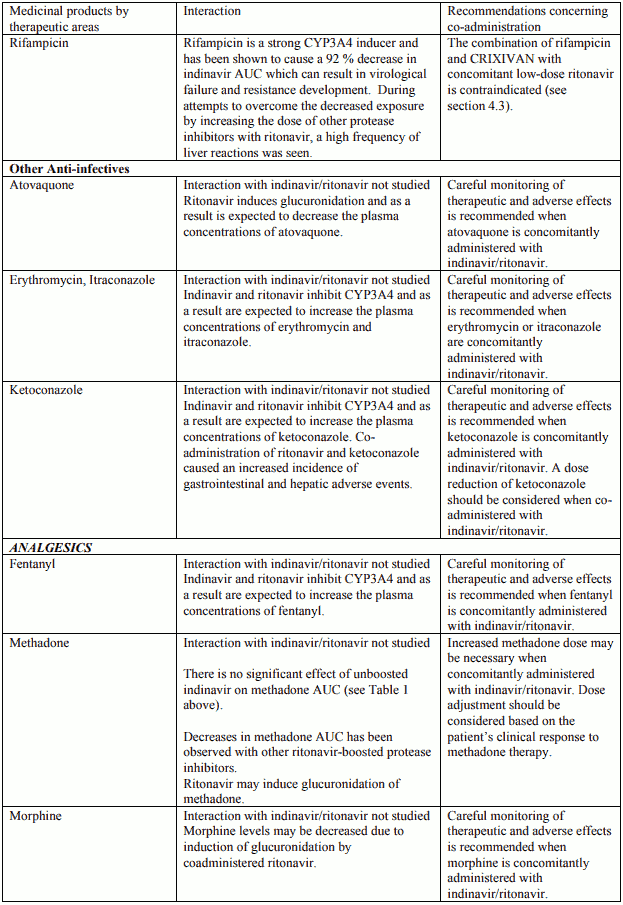
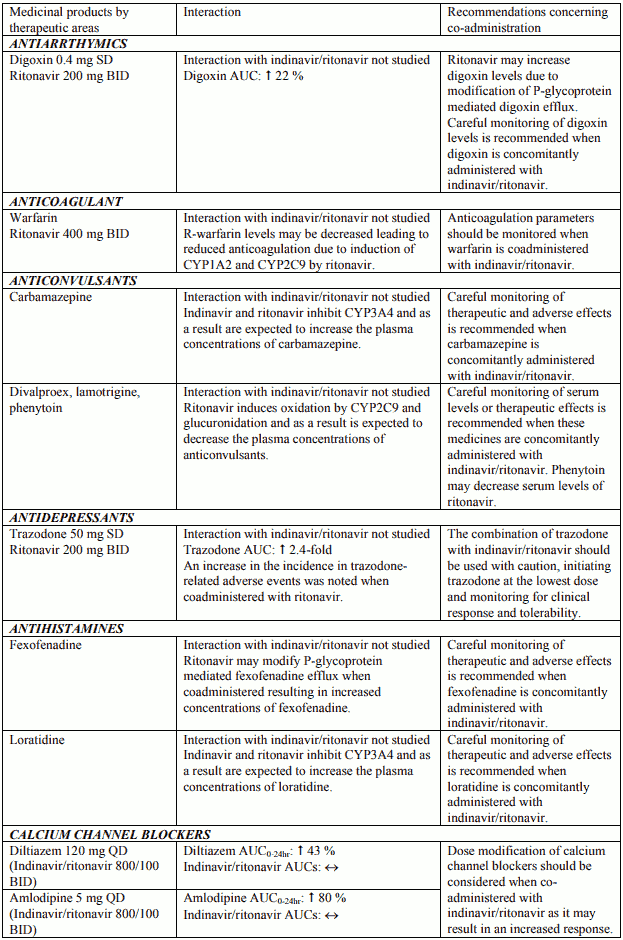
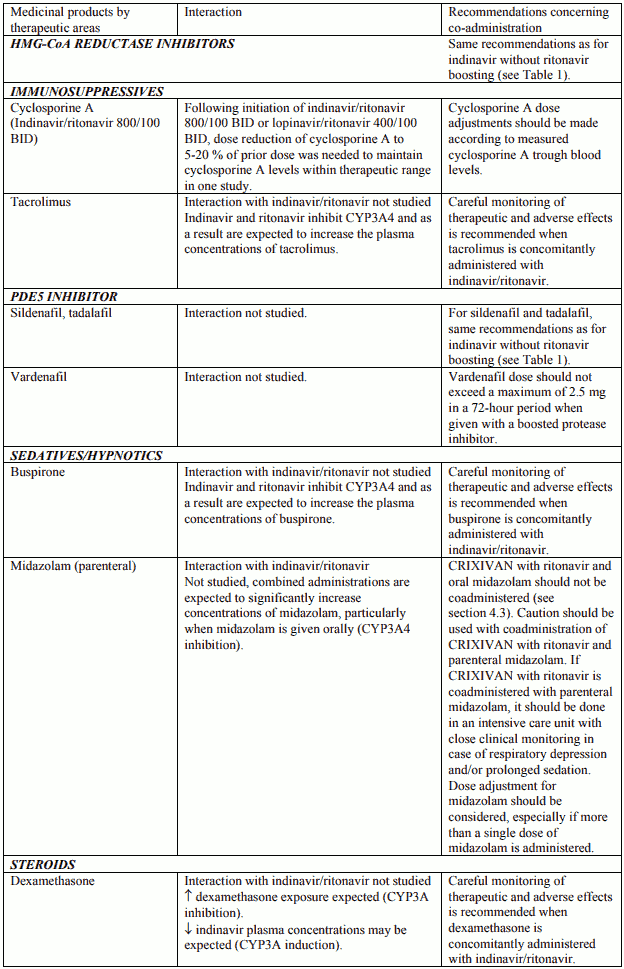
Table 2. Interactions and dose recommendations with other medical products – INDINAVIR BOOSTED WITH RITONAVIR. No specific interaction studies have been performed with the boosted dose 400 mg indinavir with 100 mg ritonavir:
Interactions between indinavir/ritonavir and other medicinal products are listed in the tables below (increase is indicated as “↑”, decrease as “↓”, no change (≤ +/- 20%) as “↔”, single dose as “SD”, once daily as “QD”, twice daily as “BID”, three times daily as “TID”, and four times daily as “QID”).
For information regarding diet or the effect of food on indinavir absorption (see sections 4.2 and 5.2).
Fertility, pregnancy and lactation
Pregnancy
There are no adequate and well-controlled studies in pregnant patients. Indinavir should be used during pregnancy only if the potential benefit justifies the potential risk to the foetus. Given that substantially lower antepartum exposures have been observed in a small study of HIV-infected pregnant patients and the limited data in this patient population, indinavir use is not recommended in HIV-infected pregnant patients (see section 5.2).
Hyperbilirubinaemia, reported predominantly as elevated indirect bilirubin, has occurred in 14% of patients during treatment with indinavir. Because it is unknown whether indinavir will exacerbate physiologic hyperbilirubinaemia in neonates, careful consideration must be given to the use of indinavir in pregnant women at the time of delivery (see section 4.8).
In Rhesus monkeys, administration of indinavir to neonates caused a mild exacerbation of the transient physiologic hyperbilirubinaemia seen in this species after birth. Administration of indinavir to pregnant Rhesus monkeys during the third trimester did not cause a similar exacerbation in neonates; however, only limited placental transfer of indinavir occurred.
Breast-feeding
It is recommended that HIVinfected women do not breastfeed their infants under any circumstances in order to avoid transmission of HIV. It is not known whether indinavir is excreted in human milk. Mothers should be instructed to discontinue breastfeeding during treatment.
Fertility
There are no data available regarding potential effects of CRIXIVAN treatment on male or female fertility.
Effects on ability to drive and use machines
No studies on the effects on the ability to drive and use machines have been performed. There are no data to suggest that indinavir affects the ability to drive and use machines. However, patients should be informed that dizziness and blurred vision have been reported during treatment with indinavir.
Undesirable effects
Nephrolithiasis occurred in approximately 10% of patients treated with the recommended (unboosted) dose of CRIXIVAN in a pooled analysis of controlled clinical trials (see also below table and in section 4.4).
Clinical adverse reactions reported by the investigators as possibly, probably, or definitely related to CRIXIVAN in ≥5% of patients treated with CRIXIVAN monotherapy or in combination with NRTI (n=309) for 24 weeks are listed below. Many of these adverse reactions were also identified as common pre-existing or frequently occurring medical conditions in this population. These adverse reactions were: nausea (35.3%), headache (25.2%), diarrhoea (24.6%), asthenia/fatigue (24.3%), rash (19.1%), taste perversion (19.1%), dry skin (16.2%), abdominal pain (14.6%), vomiting (11.0%), dizziness (10.7%). With the exception of dry skin, rash, and taste perversion, the incidence of clinical adverse reactions was similar or higher among patients treated with antiretroviral nucleoside analogue controls than among patients treated with CRIXIVAN monotherapy or in combination with NRTI. This overall safety profile remained similar for 107 patients treated with CRIXIVAN monotherapy or in combination with NRTI for up to 48 weeks. Adverse reactions, including nephrolithiasis, may lead to treatment interruption.
In controlled clinical trials conducted world-wide, indinavir was administered alone or in combination with other antiretroviral agents (zidovudine, didanosine, stavudine, and/or lamivudine) to approximately 2,000 patients, the majority of whom were adult Caucasian males (15% females).
Indinavir did not alter the type, frequency, or severity of known major adverse reactions associated with the use of zidovudine, didanosine, or lamivudine.
The following adverse reactions have been reported during clinical studies in adults and/or postmarketing use for CRIXIVAN monotherapy and/or CRIXIVAN with combination antiretroviral therapy (CART).
Very common (≥1/10); Common (≥1/100 to 1/10); Uncommon (≥1/1,000 to < 1/100); Rare (≥1/10,000 to 1/1,000); Very rare (<1/10,000); not known (cannot be estimated from the available data). Adverse reactions have also been reported during post-marketing experience* as they are derived from spontaneous reports, incidences cannot be determined.
Blood and lymphatic system disorders
Very common: increases in MCV, decreases in neutrophils
Not known*: increased spontaneous bleeding in patients with haemophilia, anemia including acute haemolytic anaemia, thrombocytopenia (see section 4.4).
Immune system disorders
Not known*: anaphylactoid reactions
Metabolism and nutrition disorders
Not known*: new onset diabetes mellitus or hyperglycaemia, or exacerbation of pre-existing diabetes mellitus, hypertriglyceridaemia, hypercholesterolaemia.
Nervous system disorders
Very common: headache, dizziness
Common: insomnia, hypoaesthesia; paraesthesia
Not known*: oral paraesthesia.
Gastrointestinal disorders
Very common: nausea, vomiting, diarrhoea, dyspepsia
Common: flatulence, dry mouth, acid regurgitation
Not known*: hepatitis, including reports of hepatic failure, pancreatitis.
Hepato-biliary disorders
Very Common: isolated asymptomatic hyperbilirubinaemia, increased ALT and AST
Not known*: liver function abnormalities
Skin and subcutaneous tissue disorders
Very common: rash, dry skin
Common: pruritus
Not known*: rash including erythema multiforme and Stevens Johnson syndrome, hypersensitivity vasculitis, alopecia, hyperpigmentation, urticaria; ingrown toenails and/or paronychia.
Musculoskeletal and connective tissue disorders
Common: myalgia
Not known*: myositis, rhabdomyolysis, increased CPK, osteonecrosis (see section 4.4), periarthritis.
Renal and urinary disorders
Very common: haematuria, proteinuria, crystalluria
Common: nephrolithiasis, dysuria.
Not known*: nephrolithiasis, in some cases with renal insufficiency or acute renal failure; pyelonephritis, interstitial nephritis, sometimes associated with indinavir crystal deposits. In some patients, resolution of the interstitial nephritis did not occur following discontinuation of indinavir therapy; renal insufficiency, renal failure, leucocyturia (see section 4.4).
General disorders and administration site conditions
Very common: asthenia/fatigue, taste perversion, abdominal pain.
Metabolic parameters
Weight and levels of blood lipids and glucose may increase during antiretroviral therapy (see section 4.4).
In HIV-infected patients with severe immune deficiency at the time of initiation of combination antiretroviral therapy (CART), an inflammatory reaction to asymptomatic or residual opportunistic infections may arise. Autoimmune disorders (such as Graves' disease and autoimmune hepatitis) have also been reported; however, the reported time to onset is more variable and these events can occur many months after initiation of treatment (see section 4.4).
Description of selected adverse reactions
Nephrolithiasis
Nephrolithiasis, including flank pain with or without haematuria (including microscopic haematuria), has been reported in approximately 10% (252/2,577) of patients receiving CRIXIVAN in clinical trials at the recommended dose compared to 2.2% in the control arms. In general, these events were not associated with renal dysfunction and resolved with hydration and temporary interruption of therapy (e.g., 1-3 days).
Hyperbilirubinaemia
Isolated asymptomatic hyperbilirubinaemia (total bilirubin ≥2.5 mg/dL, 43 mcmol/L) was reported predominantly as elevated indirect bilirubin and rarely associated with elevations in ALT, AST, or alkaline phosphatase, has occurred in approximately 14% of patients treated with CRIXIVAN alone or in combination with other antiretroviral agents. Most patients continued treatment with CRIXIVAN without dose reduction and bilirubin values gradually declined toward baseline. Hyperbilirubinaemia occurred more frequently at doses exceeding 2.4 g/day compared to doses less than 2.4 g/day.
Paediatric population
In clinical trials in paediatric patients (≥3 years), the adverse reaction profile was similar to that for adult patients except for a higher frequency of nephrolithiasis of 29% (20/70) in paediatric patients treated with CRIXIVAN. Asymptomatic pyuria of unknown etiology was noted in 10.9% (6/55) of paediatric patients who received CRIXIVAN. Some of these events were associated with mild elevation of serum creatinine.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed in Appendix V.
Incompatibilities
Not applicable.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.