Source: European Medicines Agency (EU) Revision Year: 2020 Publisher: Eli Lilly Nederland B.V., Papendorpseweg 83, 3528 BJ Utrecht, The Netherlands
Cyramza in combination with paclitaxel is indicated for the treatment of adult patients with advanced gastric cancer or gastro-oesophageal junction adenocarcinoma with disease progression after prior platinum and fluoropyrimidine chemotherapy (see section 5.1).
Cyramza monotherapy is indicated for the treatment of adult patients with advanced gastric cancer or gastro-oesophageal junction adenocarcinoma with disease progression after prior platinum or fluoropyrimidine chemotherapy, for whom treatment in combination with paclitaxel is not appropriate (see section 5.1).
Cyramza, in combination with FOLFIRI (irinotecan, folinic acid, and 5-fluorouracil), is indicated for the treatment of adult patients with metastatic colorectal cancer (mCRC) with disease progression on or after prior therapy with bevacizumab, oxaliplatin and a fluoropyrimidine.
Cyramza in combination with erlotinib is indicated for the first-line treatment of adult patients with metastatic non-small cell lung cancer with activating epidermal growth factor receptor (EGFR) mutations (see section 5.1).
Cyramza in combination with docetaxel is indicated for the treatment of adult patients with locally advanced or metastatic non-small cell lung cancer with disease progression after platinum-based chemotherapy.
Cyramza monotherapy is indicated for the treatment of adult patients with advanced or unresectable hepatocellular carcinoma who have a serum alpha fetoprotein (AFP) of ≥ 400 ng/ml and who have been previously treated with sorafenib.
Ramucirumab therapy must be initiated and supervised by physicians experienced in oncology.
The recommended dose of ramucirumab is 8 mg/kg on days 1 and 15 of a 28 day cycle, prior to paclitaxel infusion. The recommended dose of paclitaxel is 80 mg/m 2 administered by intravenous infusion over approximately 60 minutes on days 1, 8 and 15 of a 28 day cycle. Prior to each paclitaxel infusion, patients should have a complete blood count and blood chemistry performed to evaluate hepatic function. Criteria to be met prior to each paclitaxel infusion are provided in Table 1.
Table 1. Criteria to be met prior to each paclitaxel administration:
| Criteria | |
|---|---|
| Neutrophils | Day 1: ≥1.5 × 109/l, Days 8 and 15: ≥1.0 × 109/l |
| Platelets | Day 1: ≥100 × 109/l, Days 8 and 15: ≥75 × 109/l |
| Bilirubin | ≤1.5 x upper limit of normal value (ULN) |
| Aspartate aminotransferase (AST) / Alanine aminotransferase (ALT) | No liver metastases: ALT/AST ≤3 x ULN, Liver metastases: ALT/AST ≤5 x ULN |
The recommended dose of ramucirumab as a single agent is 8 mg/kg every 2 weeks.
The recommended dose of ramucirumab is 8 mg/kg every 2 weeks administered by intravenous infusion, prior to FOLFIRI administration. Prior to chemotherapy, patients should have a complete blood count. Criteria to be met prior to FOLFIRI are provided in Table 2.
Table 2. Criteria to be met prior to FOLFIRI administration:
| Criteria | |
|---|---|
| Neutrophils | ≥1.5 × 109/l |
| Platelets | ≥100 × 109/l |
| Chemotherapy-related gastro-intestinal toxicity | ≤ Grade 1 (National Cancer Institute Common Terminology Criteria for Adverse Events [NCI CTCAE]) |
The recommended dose of ramucirumab in combination with erlotinib is 10 mg/kg every two weeks.
EGFR mutation status should be determined prior to initiation of treatment with ramucirumab and erlotinib using a validated test method. See erlotinib prescribing information for the posology and method of administration of erlotinib.
The recommended dose of ramucirumab is 10 mg/kg on day 1 of a 21 day cycle, prior to docetaxel infusion. The recommended dose of docetaxel is 75 mg/m 2 administered by intravenous infusion over approximately 60 minutes on day 1 of a 21 day cycle. For East Asian patients, a reduced docetaxel starting dose of 60 mg/m 2 on day 1 of a 21 day cycle should be considered. See docetaxel prescribing information for specific dosing advice.
The recommended dose of ramucirumab as a single agent is 8 mg/kg every 2 weeks.
Patients with HCC should be selected based on a serum AFP concentration of ≥ 400 ng/ml with a validated AFP test prior to ramucirumab treatment (see section 5.1).
It is recommended that treatment be continued until disease progression or until unacceptable toxicity has occurred.
Premedication is recommended with a histamine H1 antagonist (for example diphenhydramine) prior to infusion of ramucirumab. If a patient experiences a Grade 1 or 2 infusion-related reaction premedication must be given for all subsequent infusions. If a patient experiences a second Grade 1 or 2 infusion-related reaction (IRR) administer dexamethasone (or equivalent); then, for subsequent infusions, premedicate with the following or equivalent medicinal products: an intravenous histamine H1 antagonist (for example diphenhydramine hydrochloride), paracetamol and dexamethasone.
See prescribing information for paclitaxel, for components of FOLFIRI and for docetaxel, as applicable, for premedication requirements and additional information.
The infusion rate of ramucirumab should be reduced by 50% for the duration of the infusion and all subsequent infusions if the patient experiences a grade 1 or 2 IRR. Ramucirumab should be immediately and permanently discontinued in the event of a grade 3 or 4 IRR (see section 4.4).
The blood pressure of patients should be monitored prior to each ramucirumab administration and treated as clinically indicated. Ramucirumab therapy should be temporarily discontinued in the event of severe hypertension, until controlled with medical management. If there is medically significant hypertension that cannot be controlled safely with antihypertensive therapy, ramucirumab therapy should be permanently discontinued (see section 4.4).
Patients should be monitored for the development or worsening of proteinuria during ramucirumab therapy. If the urine protein is ≥2+ on a dipstick, a 24 hour urine collection should be performed. Ramucirumab therapy should be temporarily discontinued if the urine protein level is ≥2 g/24 hours. Once the urine protein level returns to <2 g/24 hours, treatment should be resumed at a reduced dose level (see Table 3). A second dose reduction (see Table 3) is recommended if a urine protein level ≥2 g/24 hours reoccurs.
Ramucirumab therapy should be permanently discontinued if the urine protein level is >3 g/24 hours or in the event of nephrotic syndrome.
Table 3. Ramucirumab dose reductions for proteinuria:
| Initial ramucirumab dose | First dose reduction to | Second dose reduction to |
|---|---|---|
| 8 mg/kg | 6 mg/kg | 5 mg/kg |
| 10 mg/kg | 8 mg/kg | 6 mg/kg |
Ramucirumab therapy should be temporarily discontinued for at least 4 weeks prior to elective surgery. Ramucirumab therapy should be temporarily discontinued if there are wound healing complications, until the wound is fully healed (see section 4.4).
Ramucirumab therapy should be permanently discontinued in the event of:
Paclitaxel dose reductions may be applied based upon the grade of toxicity experienced by the patient. For NCI CTCAE Grade 4 haematological toxicity or Grade 3 paclitaxel-related non-haematological toxicity, it is recommended to reduce the paclitaxel dose by 10 mg/m² for all following cycles. A second reduction of 10 mg/m² is recommended if these toxicities persist or reoccur.
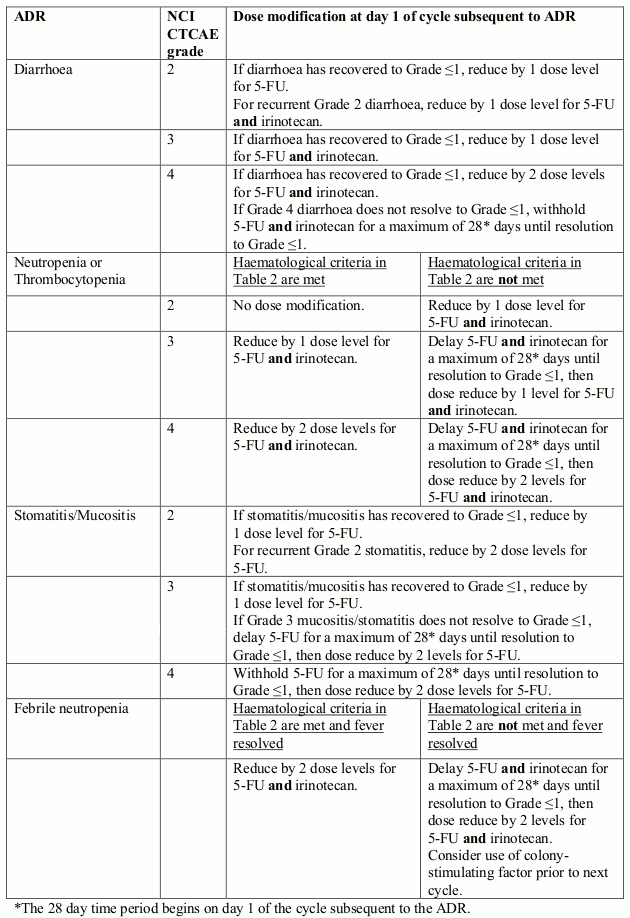
Dose reductions for individual components of FOLFIRI may be made for specific toxicities. Dose modifications of each component of FOLFIRI should be made independently and are provided in Table 4. Table 5 provides details of dose delays or dose reductions of components of FOLFIRI at the next cycle based on maximum grade of specific adverse drug reactions.
Table 4. FOLFIRI dose reductions:
| FOLFIRI componenta | Dose level | |||
|---|---|---|---|---|
| Initial dose | -1 | -2 | -3 | |
| Irinotecan | 180 mg/m2 | 150 mg/m2 | 120 mg/m2 | 100 mg/m2 |
| 5-FU bolus | 400 mg/m2 | 200 mg/m2 | 0 mg/m2 | 0 mg/m2 |
| 5-FU infusion | 2,400 mg/m2 over 46-48 hours | 2,000 mg/m2 over 46-48 hours | 1,600 mg/m2 over 46-48 hours | 1,200 mg/m2 over 46-48 hours |
a 5-FU = 5-fluorouracil.
Table 5. Dose modification of FOLFIRI components due to specific ADRs:
Docetaxel dose reductions may be applied based upon the grade of toxicity experienced by the patient. Patients who experience either febrile neutropenia, neutrophils <500 cells/mm³ for more than 1 week, severe or cumulative cutaneous reactions, or other Grade 3 or 4 non-haematological toxicities during docetaxel treatment should have treatment withheld until resolution of the toxicity. It is recommended to reduce the docetaxel dose by 10 mg/m² for all following cycles. A second reduction of 15 mg/m² is recommended if these toxicities persist or reoccur. In this case, East Asian patients with a starting dose of 60 mg/m² should have docetaxel treatment discontinued (see Posology).
In the pivotal studies there is limited evidence that patients 65 years of age or older are at increased risk of adverse events compared to patients younger than 65 years old. No dose reductions are recommended (see sections 4.4 and 5.1).
There have been no formal studies with Cyramza in patients with renal impairment. Clinical data suggest that no dose adjustments are required in patients with mild, moderate or severe renal impairment (see sections 4.4 and 5.2). No dose reductions are recommended.
There have been no formal studies with Cyramza in patients with hepatic impairment. Clinical data suggest that no dose adjustments are required in patients with mild or moderate hepatic impairment. There are no data regarding ramucirumab administration in patients with severe hepatic impairment (see sections 4.4 and 5.2). No dose reductions are recommended.
The safety and efficacy of Cyramza in children and adolescents (<18 years) has not been established. No data are available.
There is no relevant use of ramucirumab in the paediatric population for the indications of advanced gastric cancer or gastro-oesophageal adenocarcinoma, adenocarcinoma of the colon and rectum, lung carcinoma, and hepatocellular carcinoma.
Cyramza is for intravenous use. After dilution, Cyramza is administered as an intravenous infusion over approximately 60 minutes. It should not be administered as an intravenous bolus or push. To achieve the required infusion duration of approximately 60 minutes, the maximum infusion rate of 25 mg/minute should not be exceeded, instead the infusion duration should be increased. The patient should be monitored during infusion for signs of infusion-related reactions (see section 4.4) and the availability of appropriate resuscitation equipment should be ensured.
For instructions on dilution of the medicinal product before administration, see section 6.6.
There is no data on overdose in humans. Cyramza has been administered in a Phase 1 study up to 10 mg/kg every two weeks without reaching a maximum tolerated dose. In case of overdose, supportive therapy should be used.
Unopened vial: 3 years.
After dilution: When prepared as directed, infusion solutions of Cyramza contain no antimicrobial preservatives.
Chemical and physical in-use stability of Cyramza in sodium chloride 9 mg/ml (0.9%) solution for injection has been demonstrated for 24 hours at 2°C to 8°C or for 4 hours at 25°C. From a microbiological point of view, the product should be used immediately. If not used immediately, in-use storage times and conditions prior to use are the responsibility of the user and would normally not be longer than 24 hours at 2°C to 8°C, unless dilution has taken place in controlled and validated aseptic conditions.
Store in a refrigerator (2°C-8°C).
Do not freeze.
Keep the vial in the outer carton in order to protect from light.
For storage conditions after dilution of the medicinal product, see section 6.3.
10 ml solution in a vial (Type I glass) with a chlorobutyl rubber stopper, an aluminium seal and a polypropylene cap.
50 ml solution in a vial (Type I glass) with a chlorobutyl rubber stopper, an aluminium seal and a polypropylene cap.
Pack of 1 vial of 10 ml.
Pack of 2 vials of 10 ml.
Pack of 1 vial of 50 ml.
Not all pack sizes may be marketed.
Do not shake the vial.
Prepare the infusion solution using aseptic technique to ensure the sterility of the prepared solution.
Each vial is intended for single use only. Inspect the content of the vials for particulate matter and discolouration (the concentrate for solution for infusion should be clear to slightly opalescent and colourless to slightly yellow without visible particles) prior to dilution. If particulate matter or discolouration is identified, discard the vial.
Calculate the dose and volume of ramucirumab needed to prepare the infusion solution. Vials contain either 100 mg or 500 mg as a 10 mg/ml solution of ramucirumab. Only use sodium chloride 9 mg/ml (0.9%) solution for injection as a diluent.
In case of pre-filled intravenous infusion container usage: Based on the calculated volume of ramucirumab, remove the corresponding volume of sodium chloride 9 mg/ml (0.9%) solution for injection from the pre-filled 250 ml intravenous container. Aseptically transfer the calculated volume of ramucirumab to the intravenous container. The final total volume in the container should be 250 ml. The container should be gently inverted to ensure adequate mixing. Do not freeze or shake the infusion solution. Do not dilute with other solutions or co-infuse with other electrolytes or medicinal products.
In case of empty intravenous infusion container usage: Aseptically transfer the calculated volume of ramucirumab into an empty intravenous infusion container. Add a sufficient quantity of sodium chloride 9 mg/ml (0.9%) solution for injection to the container to make the total volume 250 ml. The container should be gently inverted to ensure adequate mixing. Do not freeze or shake the infusion solution. Do not dilute with other solutions or co-infuse with other electrolytes or medicinal products.
Parenteral medicinal products should be inspected visually for particulate matter prior to administration. If particulate matter is identified, discard the infusion solution.
Discard any unused portion of ramucirumab left in a vial, as the product contains no antimicrobial preservatives.
Administer via infusion pump. A separate infusion line with a protein sparing 0.22 micron filter must be used for the infusion and the line must be flushed with sodium chloride 9 mg/ml (0.9%) solution for injection at the end of the infusion.
Any unused medicinal product or waste material should be disposed of in accordance with local requirements.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.