DALIRESP Tablet Ref.[28038] Active ingredients: Roflumilast
Source: FDA, National Drug Code (US) Revision Year: 2020
12.1. Mechanism of Action
Roflumilast and its active metabolite (roflumilast N-oxide) are selective inhibitors of phosphodiesterase 4 (PDE4). Roflumilast and roflumilast N-oxide inhibition of PDE4 (a major cyclic-3′,5′-adenosine monophosphate (cyclic AMP)-metabolizing enzyme in lung tissue) activity leads to accumulation of intracellular cyclic AMP. While the specific mechanism(s) by which DALIRESP exerts its therapeutic action in COPD patients is not well defined, it is thought to be related to the effects of increased intracellular cyclic AMP in lung cells.
12.2. Pharmacodynamics
In COPD patients, 4‑week treatment with DALIRESP 500 mcg oral once daily reduced sputum neutrophils and eosinophils by 31%, and 42%, respectively. In a pharmacodynamic study in healthy volunteers, DALIRESP 500 mcg once daily reduced the number of total cells, neutrophils and eosinophils found in bronchoalveolar lavage fluid following segmental pulmonary lipopolysaccharide (LPS) challenge by 35%, 38% and 73%, respectively. The clinical significance of these findings is unknown.
12.3. Pharmacokinetics
Absorption
The absolute bioavailability of roflumilast following a 500 mcg oral dose is approximately 80%. Maximum plasma concentrations (Cmax) of roflumilast typically occur approximately one hour after dosing (ranging from 0.5 to 2 hours) in the fasted state while plateau-like maximum concentrations of the N-oxide metabolite are reached in approximately eight hours (ranging from 4 to 13 hours). Food has no effect on total drug absorption, but delays time to maximum concentration (Tmax) of roflumilast by one hour and reduces Cmax by approximately 40%, however, Cmax and Tmax of roflumilast N-oxide are unaffected. An in vitro study showed that roflumilast and roflumilast N-oxide did not inhibit P-gp transporter.
Distribution
Plasma protein binding of roflumilast and its N-oxide metabolite is approximately 99% and 97%, respectively. Volume of distribution for single‑dose 500 mcg roflumilast is about 2.9 L/kg. Studies in rats with radiolabeled roflumilast indicate low penetration across the blood-brain barrier.
Metabolism
Roflumilast is extensively metabolized via Phase I (cytochrome P450) and Phase II (conjugation) reactions. The N-oxide metabolite is the only major metabolite observed in the plasma of humans. Together, roflumilast and roflumilast N-oxide account for the majority (87.5%) of total dose administered in plasma. In urine, roflumilast was not detectable while roflumilast N-oxide was only a trace metabolite (less than 1%). Other conjugated metabolites such as roflumilast N-oxide glucuronide and 4-amino-3,5-dichloropyridine N-oxide were detected in urine.
While roflumilast is three times more potent than roflumilast N-oxide at inhibition of the PDE4 enzyme in vitro, the plasma AUC of roflumilast N-oxide on average is about 10-fold greater than the plasma AUC of roflumilast.
In vitro studies and clinical drug-drug interaction studies suggest that the biotransformation of roflumilast to its N-oxide metabolite is mediated by CYP1A2 and 3A4. Based on further in vitro results in human liver microsomes, therapeutic plasma concentrations of roflumilast and roflumilast N-oxide do not inhibit CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4/5, or 4A9/11. Therefore, there is a low probability of relevant interactions with substances metabolized by these P450 enzymes. In addition, in vitro studies demonstrated no induction of the CYP1A2, 2A6, 2C9, 2C19, or 3A4/5 and only a weak induction of CYP2B6 by roflumilast.
Elimination
The plasma clearance after short-term intravenous infusion of roflumilast is on average about 9.6 L/h. Following an oral dose, the median plasma effective half-life of roflumilast and its N-oxide metabolite are approximately 17 and 30 hours, respectively. Steady state plasma concentrations of roflumilast and its N-oxide metabolite are reached after approximately 4 days for roflumilast and 6 days for roflumilast N-oxide following once‑daily dosing. Following intravenous or oral administration of radiolabeled roflumilast, about 70% of the radioactivity was recovered in the urine.
Special Populations
Hepatic Impairment
Roflumilast 250 mcg once daily for 14 days was studied in subjects with mild-to-moderate hepatic impairment classified as Child-Pugh A and B (8 subjects in each group). The AUC of roflumilast and roflumilast N-oxide were increased by 51% and 24%, respectively in Child-Pugh A subjects and by 92% and 41%, respectively, in Child-Pugh B subjects, as compared to age-, weight-, and gender-matched healthy subjects. The Cmax of roflumilast and roflumilast N-oxide were increased by 3% and 26%, respectively, in Child-Pugh A subjects and by 26% and 40%, respectively in Child-Pugh B subjects, as compared to healthy subjects. DALIRESP 500 mcg has not been studied in hepatically impaired patients. Clinicians should consider the risk-benefit of administering DALIRESP to patients who have mild liver impairment (Child-Pugh A). DALIRESP is not recommended for use in patients with moderate or severe liver impairment (Child-Pugh B or C) [see Contraindications (4) and Use in Specific Populations (8.6)].
Renal Impairment
In twelve subjects with severe renal impairment administered a single dose of 500 mcg roflumilast, roflumilast and roflumilast N-oxide AUCs were decreased by 21% and 7%, respectively and Cmax were reduced by 16% and 12%, respectively. No dosage adjustment is necessary for patients with renal impairment [see Use in Specific Populations (8.7)].
Age
Roflumilast 500 mcg once daily for 15 days was studied in young, middle aged, and elderly healthy subjects. The exposure in elderly (>65 years of age) were 27% higher in AUC and 16% higher in Cmax for roflumilast and 19% higher in AUC and 13% higher in Cmax for roflumilast-N-oxide than that in young volunteers (18-45 years old). No dosage adjustment is necessary for elderly patients [see Use in Specific Populations (8.5)].
Gender
In a Phase I study evaluating the effect of age and gender on the pharmacokinetics of roflumilast and roflumilast N-oxide, a 39% and 33% increase in roflumilast and roflumilast N-oxide AUC were noted in healthy female subjects as compared to healthy male subjects. No dosage adjustment is necessary based on gender.
Smoking
The pharmacokinetics of roflumilast and roflumilast N-oxide were comparable in smokers as compared to non-smokers. There was no difference in Cmax between smokers and non-smokers when roflumilast 500 mcg was administered as a single dose to 12 smokers and 12 non-smokers. The AUC of roflumilast in smokers was 13% less than that in non-smokers while the AUC of roflumilast N-oxide in smokers was 17% more than that in non-smokers.
Race
As compared to Caucasians, African Americans, Hispanics, and Japanese showed 16%, 41%, and 15% higher AUC, respectively, for roflumilast and 43%, 27%, and 16% higher AUC, respectively, for roflumilast N-oxide. As compared to Caucasians, African Americans, Hispanics, and Japanese showed 8%, 21%, and 5% higher Cmax, respectively, for roflumilast and 43%, 27%, and 17% higher Cmax, respectively, for roflumilast N-oxide. No dosage adjustment is necessary for race.
Drug Interactions
Drug interaction studies were performed with roflumilast and other drugs likely to be coadministered or drugs commonly used as probes for pharmacokinetic interaction [see Drug Interactions (7)]. No significant drug interactions were observed when 500 mcg oral roflumilast was administered with inhaled salbutamol, formoterol, budesonide and oral montelukast, digoxin, theophylline, warfarin, sildenafil, midazolam, or antacids.
The effect of concomitant drugs on the exposure of roflumilast and roflumilast N-oxide is shown in the Figure 1 below.
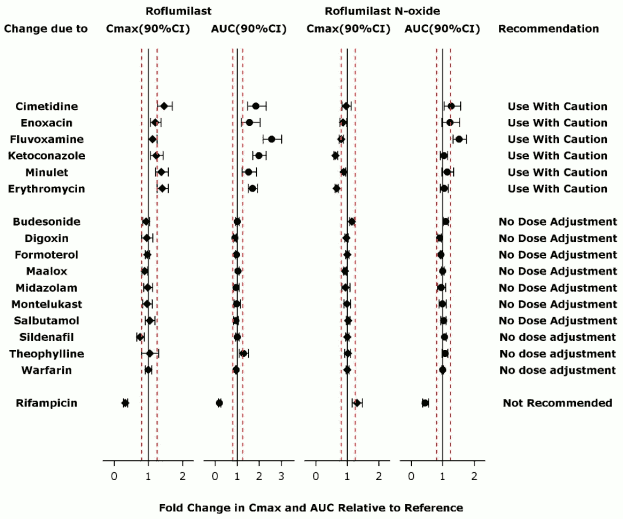
Figure 1. Effect of concomitant drugs on the exposure of roflumilast and roflumilast N-oxide. Note that the dashed lines indicate the lower and higher bounds (0.8-1.25) of the 90% confidence interval of the geometric mean ratio of Cmax or AUC for roflumilast or roflumilast N-oxide for Treatment (DALIRESP+Coadministered Drug) vs. Reference (DALIRESP). The dosing regimens of coadministered drugs was: Midazolam: 2 mg po SD; Erythromycin: 500 mg po TID; Ketoconazole: 200 mg po BID; Rifampicin: 600 mg po QD; Fluvoxamine: 50 mg po QD; Digoxin: 250 mcg po SD; Maalox: 30 mL po SD; Salbutamol: 0.2 mg po TID; Cimetidine: 400 mg po BID; Formoterol: 40 mcg po BID; Budesonide: 400 mcg po BID; Theophylline: 375 mg po BID; Warfarin: 250 mg po SD; Enoxacin: 400 mg po BID; Sildenafil: 100 mg SD; Minulet (combination oral contraceptive): 0.075 mg gestodene/0.03 mg ethinylestradiol po QD; Montelukast: 10 mg po QD
Drug interactions considered to be significant are described in more detail below [see Warnings and Precautions (5.4) and Drug Interactions (7)].
Inhibitors of CYP3A4 and CYP1A2
Erythromycin: In an open-label crossover study in 16 healthy volunteers, the coadministration of CYP3A4 inhibitor erythromycin (500 mg three times daily for 13 days) with a single oral dose of 500 mcg DALIRESP resulted in 40% and 70% increase in Cmax and AUC for roflumilast, respectively, and a 34% decrease and a 4% increase in Cmax and AUC for roflumilast N-oxide, respectively.
Ketoconazole: In an open-label crossover study in 16 healthy volunteers, the coadministration of a strong CYP3A4 inhibitor ketoconazole (200 mg twice daily for 13 days) with a single oral dose of 500 mcg DALIRESP resulted in 23% and 99% increase in Cmax and AUC for roflumilast, respectively, and a 38% reduction and 3% increase in Cmax and AUC for roflumilast N-oxide, respectively.
Fluvoxamine: In an open-label crossover study in 16 healthy volunteers, the coadministration of dual CYP 3A4/1A2 inhibitor fluvoxamine (50 mg daily for 14 days) with a single oral dose of 500 mcg DALIRESP showed a 12% and 156% increase in roflumilast Cmax and AUC along with a 210% decrease and 52% increase in roflumilast N-oxide Cmax and AUC, respectively.
Enoxacin: In an open-label crossover study in 16 healthy volunteers, the coadministration of dual CYP 3A4/1A2 inhibitor enoxacin (400 mg twice daily for 12 days) with a single oral dose of 500 mcg DALIRESP resulted in an increased Cmax and AUC of roflumilast by 20% and 56%, respectively. Roflumilast N-oxide Cmax was decreased by 14% while roflumilast N-oxide AUC was increased by 23%.
Cimetidine: In an open-label crossover study in 16 healthy volunteers, the coadministration of a dual CYP 3A4/1A2 inhibitor cimetidine (400 mg twice daily for 7 days) with a single dose of 500 mcg oral DALIRESP resulted in a 46% and 85% increase in roflumilast Cmax and AUC; and a 4% decrease in Cmax and 27% increase in AUC for roflumilast N-oxide, respectively.
Oral Contraceptives containing Gestodene and Ethinyl Estradiol
In an open-label crossover study in 20 healthy adult volunteers, coadministration of a single oral dose of 500 mcg DALIRESP with repeated doses of a fixed combination oral contraceptive containing 0.075 mg gestodene and 0.03 mg ethinyl estradiol to steady state caused a 38% increase and 12% decrease in Cmax of roflumilast and roflumilast N-oxide, respectively. Roflumilast and roflumilast N-oxide AUCs were increased by 51% and 14%, respectively.
Inducers of CYP enzymes
Rifampicin: In an open-label, three-period, fixed-sequence study in 15 healthy volunteers, coadministration of the strong CYP3A4 inducer rifampicin (600 mg once daily for 11 days) with a single oral dose of 500 mcg DALIRESP resulted in reduction of roflumilast Cmax and AUC by 68% and 79%, respectively; and an increase of roflumilast N-oxide Cmax by 30% and reduced roflumilast N-oxide AUC by 56%.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term studies were conducted in hamsters and mice with roflumilast to evaluate its carcinogenic potential. In 2-year oral gavage carcinogenicity studies, roflumilast treatment resulted in dose-related, statistically significant increases in the incidence of undifferentiated carcinomas of nasal epithelium in hamsters at ≥8 mg/kg/day (approximately 11 times the MRHD based on summed AUCs of roflumilast and its metabolites). The tumorigenicity of roflumilast appears to be attributed to a reactive metabolite of 4-amino-3,5-dichloro-pyridine N-oxide (ADCP N-oxide). No evidence of tumorigenicity was observed in mice at roflumilast oral doses up to 12 and 18 mg/kg/day in females and males, respectively (approximately 10 and 15 times the MRHD, respectively, based on summed AUCs of roflumilast and its metabolites).
Roflumilast tested positive in an in vivo mouse micronucleus test, but negative in the following assays: Ames test for bacterial gene mutation, in vitro chromosome aberration assay in human lymphocytes, in vitro HPRT test with V79 cells, an in vitro micronucleus test with V79 cells, DNA adduct formation assay in rat nasal mucosa, liver and testes, and in vivo mouse bone marrow chromosome aberration assay. Roflumilast N-oxide was negative in the Ames test and in vitro micronucleus test with V79 cells.
In a human spermatogenesis study, roflumilast 500 mcg had no effects on semen parameters or reproductive hormones during the 3-month treatment period and the following 3-month off-treatment period. In a fertility study, roflumilast decreased fertility rates in male rats at 1.8 mg/kg/day (approximately 29 times the MRHD on a mg/m2 basis). The male rats also showed increases in the incidence of tubular atrophy, degeneration in the testis and spermiogenic granuloma in the epididymides. No effect on rat fertility rate or male reproductive organ morphology was observed at 0.6 mg/kg/day (approximately 10 times the MRHD on a mg/m2 basis). In a female fertility study, no effect on fertility was observed up to the highest roflumilast dose of 1.5 mg/kg/day in rats (approximately 24 times the MRHD on a mg/m 2 basis).
14. Clinical Studies
14.1 Chronic Obstructive Pulmonary Disease (COPD)
The efficacy and safety of DALIRESP (roflumilast) in COPD was evaluated in 8 randomized, double-blind, controlled, parallel‑group clinical trials in 9394 adult patients (4425 receiving DALIRESP 500 mcg) 40 years of age and older with COPD. Of the 8 trials, two were placebo-controlled dose selection trials (Trials 1 and 2) of 6 months' duration that evaluated the efficacy of DALIRESP 250 mcg and 500 mcg once daily, four were placebo-controlled 1-year trials (Trials 3, 4, 5, and 6) primarily designed to evaluate the efficacy of DALIRESP on COPD exacerbations, and two were 6-month efficacy trials (Trials 7 and 8) which assessed the effect of DALIRESP as add-on therapy to a long-acting beta agonist or long-acting anti-muscarinic. The 8 trials enrolled patients with nonreversible obstructive lung disease (FEV1/FVC ≤70% and ≤12% or 200 mL improvement in FEV1 in response to 4 puffs of albuterol/salbutamol) but the severity of airflow obstruction at baseline was different among the trials. Patients enrolled in the dose selection trials had the full range of COPD severity (FEV1 30-80% predicted); median age of 63 years, 73% male, and 99% Caucasian. Patients enrolled in the four exacerbation trials had severe COPD (FEV1 ≤50% predicted); median age of 64 years, 74% male, and 90% Caucasian.
Patients enrolled in the two 6-month efficacy trials had moderate to severe COPD (FEV1 40-70% predicted); median age of 65 years, 68% male, and 97% Caucasian. COPD exacerbations and lung function (FEV1) were co-primary efficacy outcome measures in the four 1-year trials. In the two 6-month supportive efficacy trials, lung function (FEV1) alone was the primary efficacy outcome measure.
The two 6-month dose-selection efficacy trials (Trials 1 and 2) explored doses of 250 mcg and 500 mcg once daily in a total of 1929 patients (751 and 724 on DALIRESP 250 and 500 mcg, respectively). The selection of the 500 mcg dose was primarily based on nominal improvements in lung function (FEV1) over the 250 mcg dose. The once‑daily dosing regimen was primarily based on the determination of a plasma half-life of 17 hours for roflumilast and 30 hours for its active metabolite roflumilast N-oxide [see Clinical Pharmacology (12.3)].
An additional placebo-controlled 1-year trial (Trial 9) evaluated the effect of DALIRESP 500 mcg on COPD exacerbations when added to a fixed-dose combination (FDC) product containing an inhaled corticosteroid and long-acting beta agonist (ICS/LABA). At screening, patients were required to have two or more exacerbations in the previous year. This trial randomized a total of 2354 patients (1178 randomized to DALIRESP, 1176 to placebo). Approximately 60% of the patients enrolled had severe COPD (postbronchodilator FEV1 30%-50% of predicted) associated with chronic bronchitis and 39% had very severe COPD (postbronchodilator FEV1 ≤ 30% of predicted) associated with chronic bronchitis; mean age of 64 years, 69% male, and 80% Caucasian. The use of long-acting muscarinic antagonists was allowed.
Effect on Exacerbations
The effect of DALIRESP 500 mcg once daily on COPD exacerbations was evaluated in five 1-year trials (Trials 3, 4, 5, 6 and 9).
Two of the trials (Trials 3 and 4) conducted initially enrolled a population of patients with severe COPD (FEV1 ≤50% of predicted) inclusive of those with chronic bronchitis and/or emphysema who had a history of smoking of at least 10 pack years. Inhaled corticosteroids were allowed as concomitant medications and used in 61% of both DALIRESP and placebo-treated patients and short-acting beta agonists were allowed as rescue therapy. The use of long-acting beta agonists, long-acting anti-muscarinics, and theophylline were prohibited. The rate of moderate or severe COPD exacerbations was a co-primary endpoint in both trials. There was not a symptomatic definition of exacerbation in these 2 trials. Exacerbations were defined in terms of severity requiring treatment with a moderate exacerbation defined as treatment with systemic glucocorticosteroids in Trial 3 or systemic glucocorticosteroids and/or antibiotics in Trial 4 and a severe exacerbation defined as requiring hospitalizations and/or leading to death in Trial 3 or requiring hospitalization in Trial 4. The trials randomized 1176 patients (567 on DALIRESP) in Trial 3 and 1514 patients (760 on DALIRESP) in Trial 4. Both trials failed to demonstrate a significant reduction in the rate of COPD exacerbations.
Exploratory analyses of the results of Trials 3 and 4 identified a subpopulation of patients with severe COPD associated with chronic bronchitis and COPD exacerbations within the previous year that appeared to demonstrate a better response in the reduction of the rate of COPD exacerbations compared to the overall population. As a result, two subsequent trials (Trial 5 and Trial 6) were conducted that enrolled patients with severe COPD but associated with chronic bronchitis, at least one COPD exacerbation in the previous year, and at least a 20 pack-year smoking history. In these trials, long-acting beta agonists and short-acting anti-muscarinics were allowed and were used by 44% and 35% of patients treated with DALIRESP and 45% and 37% of patients treated with placebo, respectively. The use of inhaled corticosteroids was prohibited. As in trials 3 and 4, the rate of moderate exacerbations (defined as requiring intervention with systemic glucocorticosteroids) or severe exacerbations (defined as leading to hospitalization and/or to death) was a co-primary endpoint.
Trial 5 randomized a total of 1525 patients (765 on DALIRESP) and Trial 6 randomized a total of 1571 patients (772 on DALIRESP). In both trials, DALIRESP 500 mcg once daily demonstrated a significant reduction in the rate of moderate or severe exacerbations compared to placebo (Table 2). These two trials provide the evidence to support the use of DALIRESP for the reduction of COPD exacerbations.
Table 2: Effect of DALIRESP on Rate of Moderate or Severe Exacerbations
| Study | Exacerbations Per Patient-Year | |||||
| DALIRESP | Placebo | Absolute Reduction* | RR† | 95% CI | Percent Reduction‡ | |
| Trial 5 | 1.1 | 1.3 | 0.2 | 0.85 | 0.74, 0.98 | 15 |
| Trial 6 | 1.2 | 1.5 | 0.3 | 0.82 | 0.71, 0.94 | 18 |
* Absolute reduction measured as difference between placebo and roflumilast‑treated patients.
† RR is Rate Ratio.
‡ Percent reduction is defined as 100 (1-RR).
For patients in Trials 5 and 6 who received concomitant long-acting beta agonists or short-acting anti-muscarinics, reduction of moderate or severe exacerbations with DALIRESP was similar to that observed for the overall populations of the two trials.
In Trial 9, when added to background therapy of FDC ICS/LABA, the rate ratio for COPD exacerbations among patients administered DALIRESP vs. placebo was 0.92 (95% CI 0.81, 1.04).
Effect on Lung Function
While DALIRESP is not a bronchodilator, all 1-year trials (Trials 3, 4, 5, and 6) evaluated the effect of DALIRESP on lung function as determined by the difference in FEV1 between DALIRESP and placebo-treated patients (pre-bronchodilator FEV1 measured prior to study drug administration in three of the trials and post-bronchodilator FEV1 measured 30 minutes after administration of 4 puffs of albuterol/salbutamol in one trial) as a co-primary endpoint. In each of these trials DALIRESP 500 mcg once daily demonstrated a statistically significant improvement in FEV1 which averaged approximately 50 mL across the four trials. Table 3 shows FEV1 results from Trials 5 and 6 which had demonstrated a significant reduction in COPD exacerbations.
Table 3. Effect of DALIRESP on FEV1:
| Study | Change in FEV1 from Baseline, mL | |||
| DALIRESP | Placebo | Effect* | 95% CI | |
| Trial 5 | 46 | 8 | 39 | 18, 60 |
| Trial 6 | 33 | -25 | 58 | 41,75 |
* Effect measured as difference between DALIRESP and placebo treated patients.
Lung function was also evaluated in two 6-month trials (Trials 7 and 8) to assess the effect of DALIRESP when administered as add-on therapy to treatment with a long-acting beta agonist or a long-acting anti-muscarinic. These trials were conducted in a different population of COPD patients [moderate to severe COPD (FEV1 40 to 70% of predicted) without a requirement for chronic bronchitis or frequent history of exacerbations] from that for which efficacy in reduction of exacerbations has been demonstrated and provide safety support to the DALIRESP COPD program.
Starting dose titration trial
The tolerability of DALIRESP was evaluated in a 12-week randomized, double-blind, parallel group trial in patients with severe COPD associated with chronic bronchitis (Trial 10). At screening, patients were required to have had at least one exacerbation in the previous year. A total of 1323 patients were randomized to receive DALIRESP 500 mcg once a day for 12 weeks (n=443), DALIRESP 500 mcg every other day for 4 weeks followed by DALIRESP 500 mcg once a day for 8 weeks (n=439), or DALIRESP 250 mcg once a day for 4 weeks followed by DALIRESP 500 mcg once a day for 8 weeks (n=441).
Over the 12-week study period, the percentage of patients discontinuing treatment was 6.2% lower in patients initially receiving DALIRESP 250 mcg daily for 4 weeks followed by DALIRESP 500 mcg daily for 8 weeks (18.4%) compared to those receiving DALIRESP 500 mcg daily for 12 weeks (24.6%) (Odds Ratio = 0.66; 95% CI: 0.47 to 0.93; p=0.017). Because this trial was limited to 12 weeks in duration, whether initiation of dosing with DALIRESP 250 mcg improves the long term tolerability of DALIRESP 500 mcg has not been determined.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.