DATROWAY Powder for concentrate for solution for infusion Ref.[115207] Active ingredients: Datopotamab deruxtecan
Source: European Medicines Agency (EU) Revision Year: 2025 Publisher: Daiichi Sankyo Europe GmbH, Zielstattstrasse 48, 81379 Munich, Germany
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic agents, monoclonal antibodies and antibody drug conjugates
ATC code: L01FX35
Mechanism of action
Datopotamab deruxtecan is a TROP2-directed antibody-drug conjugate. The antibody is a humanised anti-TROP2 IgG1 attached to deruxtecan, a topoisomerase I inhibitor (DXd) bound by a tetrapeptide-based cleavable linker. The antibody-drug conjugate is stable in plasma. The antibody binds to TROP2 expressed on the surface of certain tumour cells. After binding, datopotamab deruxtecan undergoes internalisation into the tumour cells. Subsequently, the release of DXd results in DNA damage and apoptotic cell death via topoisomerase I inhibition. Datopotamab deruxtecan may also exhibit indirect cytotoxicity as shown in vitro through mechanisms of antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP) and bystander cytotoxicity of DXd against TROP2 expressing tumour cells and neighbouring cells.
Pharmacodynamic effects
Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity. During the median 5.5 month treatment period across clinical studies in NSCLC and breast cancer patients treated with Datroway at 6 mg/kg, the incidence of anti-datopotamab deruxtecan antibodies was 16% (146 out of 912) and the incidence of neutralising antibodies against datopotamab deruxtecan was 2.5% (23 out of 912). There was no apparent effect of anti-drug antibodies on the pharmacokinetics or effectiveness of datopotamab deruxtecan. No clinically meaningful impact on the safety of datopotamab deruxtecan was observed.
Clinical efficacy and safety
HR+/HER2- breast cancer
TROPION-Breast01 (NCT05104866)
The efficacy of Datroway was evaluated in study TROPION-Breast01, a multicentre, open-label, randomised study of 732 patients with unresectable or metastatic HR-positive, HER2-negative (IHC 0, IHC1+ or IHC2+/ISH-) breast cancer. Patients must have progressed on and been unsuitable for endocrine therapy. Patients were required to have received 1 to 2 lines of prior chemotherapy in the unresectable or metastatic disease setting. Patients with clinically inactive brain metastases were included in the study. Patients were excluded for a history of ILD/pneumonitis requiring treatment with steroids, ongoing ILD/pneumonitis, or clinically significant cardiac, pulmonary or corneal disease at screening. Patients were also excluded for Eastern Cooperative Oncology Group (ECOG) performance status >1.
A total of 732 patients were randomised 1:1 to receive either Datroway 6 mg/kg (N=365) by intravenous infusion every 3 weeks or physician's choice of chemotherapy (N=367, eribulin 59.9%, capecitabine 20.7%, vinorelbine 10.4% or gemcitabine 9.0%) until unacceptable toxicity or disease progression. Randomisation was stratified by previous lines of chemotherapy (one or two), prior treatment with a CDK4/6 inhibitor (yes or no) and geographical region (United States, Canada, Europe, or Rest of World). Tumour imaging was obtained every 6 weeks until disease progression.
The dual primary efficacy endpoints were progression-free survival (PFS) as assessed by blinded independent central review (BICR) based on Response Evaluation Criteria in Solid Tumours (RECIST) v.1.1 and overall survival (OS). Confirmed objective response rate (ORR) and duration of response (DOR) were secondary endpoints.
Baseline demographics and disease characteristics were similar between treatment arms. The median age was 55 years (range 28 to 86); 22.3% were ≥65 years and 98.8% were female; 47.8% were White, 1.5% were Black or African American, 40.7% were Asian and 11.3% were of Hispanic/Latino ethnicity; 57% had ECOG PS 0 and 42.3% had ECOG PS of 1; 97.3% had visceral disease, 71.9% had liver metastases and 7.9% had stable brain metastases at baseline at the time of randomisation.
There were 60.2% of patients who received prior endocrine therapy in the (neo) adjuvant setting, 88.5% received prior endocrine therapy in the unresectable or metastatic setting and all patients received prior chemotherapy regimens in the unresectable or metastatic setting. Overall, 80.7% of patients had received prior taxanes and 63.8% had received prior anthracyclines. There were 62% of patients who had 1 prior chemotherapy regimen and 37.7% of patients who had 2 prior chemotherapy regimens for treatment of unresectable or metastatic disease. 82.5% of patients had prior treatment with a CDK4/6 inhibitor.
Efficacy results are shown in Table 4 and Figure 1 and 2.
Table 4. Efficacy results in TROPION-Breast01:
| Efficacy parameter | Datroway (N=365) | Chemotherapy (N=367) |
|---|---|---|
| Progression-free survival by BICRa | ||
| Number of events (%) | 212 (58.1) | 235 (64.0) |
| Median, months (95% CI) | 6.9 (5.7, 7.4) | 4.9 (4.2, 5.5) |
| Hazard ratio (95% CI) | 0.63 (0.52, 0.76) | |
| p-valueb | <0.0001 | |
| Overall Survivalc,d | ||
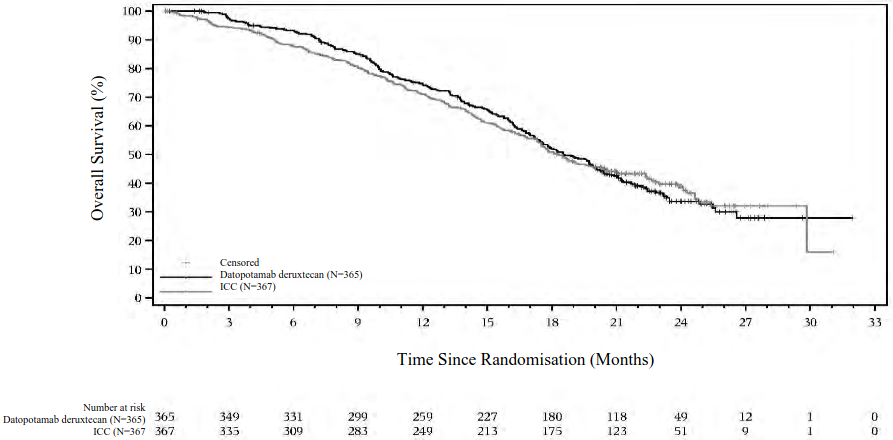
| Number of events (%) | 223 (61.1) | 213 (58.0) |
| Median, months (95% CI) | 18.6 (17.3, 20.1) | 18.3 (17.3, 20.5) |
| Hazard ratio (95% CI) | 1.01 (0.83, 1.22) | |
| p-valuee | 0.9445 | |
| Objective response rate by BICRa,f | ||
| n (%) | 133 (36.4) | 84 (22.9) |
| 95% CI | 31.4, 41.3 | 18.6, 27.2 |
| Duration of response by BICRa,f | ||
| Median, months (95% CI) | 6.7 (5.6, 9.8) | 5.7 (4.9, 6.8) |
a Data cutoff 17 July 2023
b Predefined p-value boundary was 0.01.
c Data cutoff 24 July 2024
d 12.3% and 24.0% of patients in the datopotamab deruxtecan and ICC arms, respectively, received subsequent treatment with trastuzumab deruxtecan and/or sacituzumab govitecan post discontinuation.
e Predefined p-value boundary was 0.0403.
f Endpoints were analysed descriptively.
Figure 1. Kaplan-Meier plot of PFS by BICR in TROPION-Breast01 (data cutoff 17 July 2023):
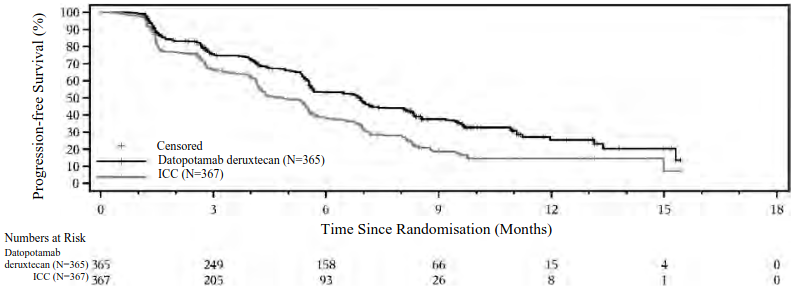
The improvement in PFS by BICR was consistent amongst the prespecified subgroups of patients including by geographic region, prior use of CDK4/6 inhibitor and previous line of therapy.
Figure 2. Kaplan-Meier plot of final OS in TROPION-Breast01 (data cutoff 24 July 2024):
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with Datroway in all subsets of the paediatric population in breast cancer (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
The pharmacokinetics of datopotamab deruxtecan was evaluated in 729 patients.
At the recommended dosage of Datroway, the geometric mean (coefficient of variation [CV]%) Cmax of datopotamab deruxtecan and DXd were 154 μg/mL (20.3%) and 2.82 ng/mL (58.1%), respectively, and the AUC (area under the plasma concentration versus time curve) of datopotamab deruxtecan and DXd were 671 μg*day/mL (31.4%) and 18.5 ng*day/mL (42.6%) after the first dose in cycle 1, respectively.
Distribution
Steady state volume of distribution of datopotamab deruxtecan is 3.52 L. In vitro, across the concentration range of 10 ng/mL to 100 ng/mL, the mean human plasma protein binding of DXd was 96.8 to 98.0%, and the blood-to-plasma concentration ratio of DXd was 0.59-0.62.
Biotransformation
Datopotamab deruxtecan undergoes intracellular cleavage by lysosomal enzymes to release DXd. The humanised TROP2 IgG1 monoclonal antibody is expected to be degraded into small peptides and amino acids via catabolic pathways in the same manner as endogenous IgG. In vitro metabolism studies in human liver microsomes indicate that DXd is primarily metabolised by CYP3A4 via oxidative pathways and does not undergo significant metabolism by UGT or other CYP enzymes.
Elimination
Following intravenous administration of datopotamab deruxtecan in patients, the clearance of datopotamab deruxtecan was estimated to be 0.57 L/day. The median elimination half-life (t1/2) of datopotamab deruxtecan was 4.82 days and apparent median t1/2 of released DXd was approximately 5.50 days. In vitro, DXd was a substrate of P-gp, OATP1B1, OATP1B3, MATE2-K, MRP1 and BCRP. DXd excretion was not studied in humans.
In vitro interactions
Effects of Datroway on the pharmacokinetics of other medicinal products
In vitro studies indicate DXd does not inhibit or induce major CYP450 enzymes including CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6 and 3A. In vitro studies indicate that DXd does not inhibit OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3, MATE1, MATE2-K, P-gp, BCRP or BSEP transporters.
Effects of other medicinal products on the pharmacokinetics of Datroway
In vitro, DXd was a substrate of P-gp, OATP1B1, OATP1B3, MATE2-K, MRP1 and BCRP. No clinically meaningful interaction is expected with medicinal products that are inhibitors of MATE2-K, MRP1, P-gp, OATP1B1 or BCRP transporters (see section 4.5).
Linearity/non-linearity
The exposure of datopotamab deruxtecan and released DXd when administered intravenously increased in proportion to dose in the 4 mg/kg to 10 mg/kg dose range (approximately 0.7 to 1.7 times the recommended dose). No accumulation of datopotamab deruxtecan was observed at the 6 mg/kg dose between cycle 1 and cycle 3.
Special populations
Based on population pharmacokinetic analysis, age (26 to 86 years), race, region, and sex did not have a clinically meaningful effect on exposure of datopotamab deruxtecan or DXd. The mean volume of distribution and clearance of datopotamab deruxtecan and DXd increase with increasing body weight (35.6 kg to 156 kg). This is considered clinically relevant. See section 4.2 for dose recommendations.
Renal impairment
No dedicated renal impairment study was conducted. Based on population pharmacokinetic analysis including patients with mild to moderate (CLcr ≥30 and <90 mL/min) renal impairment (estimated by Cockcroft-Gault), the pharmacokinetics of datopotamab deruxtecan or DXd was not affected by mild to moderate renal impairment as compared to normal renal function (CLcr ≥90 mL/min) (see section 4.2).
Hepatic impairment
No dedicated hepatic impairment study was conducted. Based on population pharmacokinetic analysis including patients with mild hepatic impairment (total bilirubin ≤ ULN and any AST > ULN or total bilirubin >1 to 1.5 times ULN and any AST), the pharmacokinetics of datopotamab deruxtecan or DXd was not affected by mild hepatic impairment as compared to normal hepatic function. There are limited data in patients with moderate (total bilirubin >1.5 to 3 times ULN and any AST) hepatic impairment to draw conclusions. Insufficient data are available for patients with severe (total bilirubin >3 times ULN and any AST) hepatic impairment. Therefore, patients with moderate and severe hepatic impairment should be monitored carefully (see section 4.2 and 4.4).
5.3. Preclinical safety data
Repeat-dose toxicity
In animals, toxicities were observed in lymphatic and haematopoietic organs, intestines, kidneys, male and female reproductive tracts, lung, skin, eye (cornea), liver and incisor teeth following the administration of datopotamab deruxtecan at exposure levels of the topoisomerase I inhibitor below clinical plasma exposure. In these animals, ADC exposure levels were similar or above clinical plasma exposure.
Genotoxicity
DXd was clastogenic in both an in vivo rat bone marrow micronucleus assay and an in vitro Chinese hamster lung chromosome aberration assay.
Carcinogenicity
Carcinogenicity studies have not been conducted with datopotamab deruxtecan.
Reproductive and developmental toxicity
Dedicated fertility studies have not been conducted with datopotamab deruxtecan. Based on the results from an animal toxicity study in rats, datopotamab deruxtecan at 200 mg/kg (approximately 29 times the human recommended dose of 6 mg/kg based on AUC) may impair male and female reproductive function and fertility at exposure levels of the topoisomerase I inhibitor below clinical plasma exposure. Toxicity to male reproductive tract included testis (degeneration of germinal epithelium and atrophy of seminiferous tubule) and epididymis (single cell necrosis of ductal epithelium, cell debris in duct and decreased number of spermatozoa in duct), which did not reverse after 8 weeks of treatment cessation, except for single cell necrosis of ductal epithelium. The effects on female fertility, including an increase in the number of atretic follicles in the ovaries and single cell necrosis of mucosal epithelium in the vagina, may be reversible.
Reproductive and developmental toxicity studies have not been conducted with datopotamab deruxtecan. Based on results from general animal toxicity studies, datopotamab deruxtecan and DXd were toxic to rapidly dividing cells (testes), and DXd was genotoxic, suggesting the potential for embryotoxicity and teratogenicity.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.