DECTOVA Solution for infusion Ref.[27672] Active ingredients: Zanamivir
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: GlaxoSmithKline Trading Services Limited, 12 Riverwalk, Citywest Business Campus, Dublin 24, Ireland
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Antivirals for systemic use, neuraminidase inhibitors
ATC code: J05AH01
Mechanism of action
Zanamivir is an inhibitor of influenza virus neuraminidase, an enzyme that releases viral particles from the plasma membrane of infected cells and promotes virus spread in the respiratory tract.
In vitro activity
Neuraminidase inhibition occurred at very low zanamivir concentrations in vitro, with median inhibitory (IC50) values of 0.33 nM to 5.77 nM against influenza A and B strains respectively.
Resistance
Resistance selection during zanamivir treatment is rare. Reduced susceptibility to zanamivir is associated with mutations that result in amino acid changes in the viral neuraminidase or viral hemagglutinin or both. Neuraminidase substitutions conferring reduced susceptibility to zanamivir have emerged during treatment with zanamivir in human viruses and those with zoonotic potential: E119D, E119G, I223R, R368G, G370D, N434S (A/H1N1); N294S, T325I (A/H3N2); R150K (B); R292K (A/H7N9). The neuraminidase substitution Q136K (A/H1N1 and A/H3N2), confers high level resistance to zanamivir but is selected during adaptation to cell culture and not during treatment.
The clinical impact of reduced susceptibility in these viruses is unknown, and the effects of specific substitutions on virus susceptibility to zanamivir may be strain-dependent.
Cross-resistance
Cross-resistance between zanamivir and oseltamivir or peramivir has been observed in neuraminidase inhibition assays. A number of neuraminidase amino acid substitutions that arise during oseltamivir or peramivir treatment result in reduced susceptibility to zanamivir. The clinical impact of substitutions associated with reduced susceptibility to zanamivir and other neuraminidase inhibitors is variable and may be strain-dependent.
The H275Y substitution is the most common neuraminidase resistance substitution and is associated with reduced susceptibility to peramivir and oseltamivir. This substitution has no effect on zanamivir; therefore, viruses with the H275Y substitution retain full susceptibility to zanamivir.
Clinical efficacy
Human challenge study
A double-blind, randomised study to examine the prophylactic antiviral activity and efficacy of repeat dose zanamivir 600 mg every 12 hours intravenously compared to placebo in healthy male volunteers against infection from inoculation with influenza A/Texas/91 (H1N1) virus was conducted. Zanamivir had a significant prophylactic effect against an experimental challenge with influenza A virus as demonstrated by the low infection rate (14% vs. 100% positive serology in placebo group, p<0.005), isolation of virus by viral culture (0% vs. 100% in placebo group, p<0.005), as well as reductions in fever (14% vs. 88% in placebo group, p <0.05), upper respiratory tract illness (0% versus 100% in placebo group, p<0.005) and total symptom scores (1 vs. 44 median score in placebo group, p<0.001).
Bronchoalveolar lavage study
A Phase I, open-label study to evaluate serum and lower respiratory pharmacokinetics following administration of intravenous and inhaled zanamivir to healthy adult subjects utilising bronchoalveolar lavage fluid was conducted. The 600 mg dose given intravenously best approximated epithelial lining fluid concentrations achieved by the approved 10 mg dose of zanamivir inhalation powder which demonstrated efficacy in large clinical studies in uncomplicated influenza.
Phase III study in patients with complicated influenza
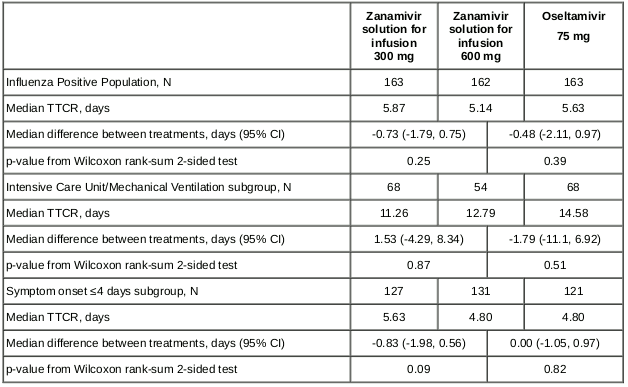
A Phase III, double-blind, study was conducted to evaluate the efficacy, antiviral activity and safety of zanamivir 600 mg twice daily intravenously compared to oral oseltamivir 75 mg twice daily and 300 mg zanamivir twice daily intravenously in hospitalised patients (>16 years of age) with influenza. The median patient age was 57 years and 35% (218/615) of patients were ≥65 years, of which 17% (n=103) were 65 to <75; 14% (n=84) were 75 to <85, and 5% (n=31) were ≥85 years of age. Patients were stratified at randomisation based on time from onset of symptoms to initiation of treatment (≤4 days and 5 to 6 days). Eligible patients were not to have had >3 days of prior antiviral treatment. The initial 5 day treatment course could be extended for up to 5 additional days if clinical symptoms or patient characteristics warranted further treatment. The primary endpoint was time to clinical response (TTCR); clinical response was defined as a composite of vital sign stabilisation (temperature, oxygen saturation, respiratory status, heart rate and systolic blood pressure) or hospital discharge. The primary analysis was performed on the Influenza Positive Population (IPP) comprised of 488 patients. The study did not meet its pre-specified primary objective of demonstrating superiority of 600 mg zanamivir to oral oseltamivir or to 300 mg zanamivir in TTCR. There were no significant differences in TTCR across treatment comparisons in the overall IPP or in two pre-specified subgroups (Table 6).
Table 6. Statistical comparisons of TTCR between the 600 mg zanamivir group and each other group (IPP):
This medicinal product has been authorised under ‘exceptional circumstances’.
This means that for scientific reasons it has not been possible to obtain complete information on this medicinal product.
The European Medicines Agency will review any new information which may become available every year and this SmPC will be updated as necessary.
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with Dectova in one or more subsets of the paediatric population in the treatment and prevention of influenza (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
The serum pharmacokinetics of zanamivir administered intravenously have been studied in healthy volunteers receiving single escalating doses from 1 to 1200 mg and repeated doses of 600 mg twice daily for 5 days. Hospitalised patients with influenza also have received 300 mg or 600 mg twice daily for 5 to 10 days.
Dose proportionality was observed in zanamivir Cmax and AUC and no accumulation of zanamivir in serum was evident after repeated intravenous doses of up to 600 mg.
Distribution
The plasma protein binding of zanamivir is very low (less than 10%). The volume of distribution of zanamivir in adults is approximately 16 litres, which approximates the volume of extracellular water.
Following twice-daily administration of zanamivir solution for infusion, pulmonary epithelial lining fluid concentrations were 60-65% of the serum concentrations at the corresponding sampling time 12 hours after dosing. Following twice daily administration of 600 mg zanamivir solution for infusion, median trough zanamivir epithelial lining fluid concentrations ranged from 419 ng/mL to 584 ng/mL and were 47-66% of those in the initial bronchoalveolar sample following orally zanamivir inhalation powder 10 mg twice daily.
In vitro studies indicate that zanamivir is not an inhibitor or substrate of Breast Cancer Resistant Protein (BCRP), P-glycoprotein, Multidrug And Toxin Extrusion protein (MATE)1, MATE2-K, Organic Anion Transporter (OAT)1, OAT3, Organic Anion Transporting Polypeptide (OATP)1B1, OATP1B3 and Organic Cation Transporter (OCT)2 transporters.
Biotransformation
There is no evidence that zanamivir is metabolised.
Zanamivir is not an inhibitor of cytochrome P450 (CYP) enzymes CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6 and 3A4. Zanamivir is not an inducer of CYP1A2 and 2B6 and, although induction of CYP3A4 in vitro was observed at 50-fold higher than the clinically relevant concentrations, no interaction with CYP3A4 substrates is expected based on physiologically based pharmacokinetic modelling.
Elimination
Zanamivir is eliminated unchanged in urine by glomerular filtration. In adults with normal renal function, the elimination half-life is approximately 2-3 hours.
Elderly
The pharmacokinetics in elderly subjects was similar to young adult subjects. In the population pharmacokinetic analysis, age had no significant effect on the pharmacokinetics of zanamivir.
Paediatric population
The pharmacokinetics of zanamivir following a twice daily intravenous dose of 14 mg/kg for paediatric patients between 6 months and <6 years and 12 mg/kg for those between 6 years and <18 years of age were similar to those seen in adults who received 600 mg twice daily intravenously. The pharmacokinetics of zanamivir in subjects 6 months to <18 years of age (administered standard dose of 12 mg/kg, 14 mg/kg or 600 mg according to age and body weight) and in adult subjects (administered standard dose of 600 mg) was similar (Table 7).
Table 7. Pharmacokinetic parameters in paediatric and adult subjects:
| Age Group | Dose | N | Cmax (μg/mL) | AUC(0-∞) (μg.h/mL) | Cmin (μg/mL) | T1/2 (h) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| GM | %CV | GM | %CV | GM | Range | GM | %CV | |||
| 6 months - <1 year | 14 mg/kg | 7 | 36.2 | 21 | 75.3 | 23 | NA | NA | 1.84 | 19 |
| 1 - <2 years | 14 mg/kg | 6 | 37.8 | 24 | 72.4 | 14 | 0.305 | NA | 2.49 | 118 |
| 2 - <6 years | 14 mg/kg | 12 | 41.5 | 23 | 80.3 | 38 | 0.277 | 0.133 – 0.984 | 1.60 | 34 |
| 6 - <13 years | 12 mg/kg | 16 | 44.2 | 47 | 107 | 41 | 0.564 | 0.111 – 2.31 | 2.57 | 55 |
| 13 - <18 years | 600 mg | 13 | 34.5 | 27 | 91.1 | 27 | 0.211 | 0.104 – 0.428 | 2.06 | 47 |
| >18 years | 600 mg | 67 | 32.8 | 34 | 82.9 | 36 | 0.82 | 0.1 – 11.4 | 2.39 | 31 |
%CV = percent coefficient of variation, GM = Geometric Mean, NA = Not available
Renal impairment
The serum half-life of zanamivir increases to approximately 12-20 hours in patients with severe renal impairment (creatinine clearance <30 mL/min). Dectova has not been studied in patients with end-stage renal disease.
There are limited data on zanamivir exposure during concomitant continuous renal replacement therapy and very limited data with dialysis.
Hepatic impairment
Zanamivir is not metabolised, therefore no effect of hepatic impairment is expected.
Race
Pharmacokinetic studies in Thai, Chinese and Japanese healthy subjects did not identify any clinically relevant differences in the pharmacokinetics of zanamivir in these populations compared with Caucasians.
5.3. Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential, or toxicity to reproduction and development, with the exception of a rat embryofoetal development study (subcutaneous administration). In the rat embryofoetal study, there was an increase in the incidence rates of a variety of minor skeletal and visceral alterations, most of which remained within the background rates of the historical occurrence in the strain studied.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.