DIMETHYL FUMARATE MYLAN Gastro-resistant hard capsule Ref.[50260] Active ingredients: Dimethyl fumarate
Source: European Medicines Agency (EU) Revision Year: 2022 Publisher: Mylan Ireland Limited, Unit 35/36 Grange Parade, Baldoyle Industrial Estate, Dublin 13, Ireland
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Immunosuppressants, other immunosuppressants
ATC code: L04AX07
Mechanism of action
The mechanism by which dimethyl fumarate exerts therapeutic effects in multiple sclerosis is not fully understood. Preclinical studies indicate that dimethyl fumarate pharmacodynamic responses appear to be primarily mediated through activation of the Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) transcriptional pathway. Dimethyl fumarate has been shown to up regulate Nrf2-dependent antioxidant genes in patients (e.g. NAD(P)H dehydrogenase, quinone 1; [NQO1]).
Pharmacodynamic effects
Effects on the immune system
In preclinical and clinical studies, dimethyl fumarate demonstrated anti-inflammatory and immunomodulatory properties. Dimethyl fumarate and monomethyl fumarate, the primary metabolite of dimethyl fumarate, significantly reduced immune cell activation and subsequent release of proinflammatory cytokines in response to inflammatory stimuli in preclinical models. In clinical studies with psoriasis patients, dimethyl fumarate affected lymphocyte phenotypes through a down-regulation of pro-inflammatory cytokine profiles (TH1, TH17), and biased towards anti-inflammatory production (TH2). Dimethyl fumarate demonstrated therapeutic activity in multiple models of inflammatory and neuroinflammatory injury. In Phase 3 studies in MS patients (DEFINE, CONFIRM and ENDORSE), upon treatment with Tecfidera mean lymphocyte counts decreased on average by approximately 30% of their baseline value over the first year with a subsequent plateau. In these studies, patients who discontinued Tecfidera therapy with lymphocyte counts below the lower limit of normal (LLN, 910 cells/mm³) were monitored for recovery of lymphocyte counts to the LLN.
Figure 1 shows the proportion of patients estimated to reach the LLN based on the Kaplan-Meier method without prolonged severe lymphopenia. The recovery baseline (RBL) was defined as the last on-treatment ALC prior to Tecfidera discontinuation. The estimated proportion of patients recovering to LLN (ALC ≥0.9 x 109/L) at Week 12 and Week 24, who had mild, moderate, or severe lymphopenia at RBL are presented in Table 1, Table 2, and Table 3 with 95% pointwise confidence intervals. The standard error of the Kaplan-Meier estimator of the survival function is computed using Greenwood's formula.
Figure 1. Kaplan-Meier Method; Proportion of Patients with Recovery to ≥910 cells/mm³ LLN from the Recovery Baseline (RBL):
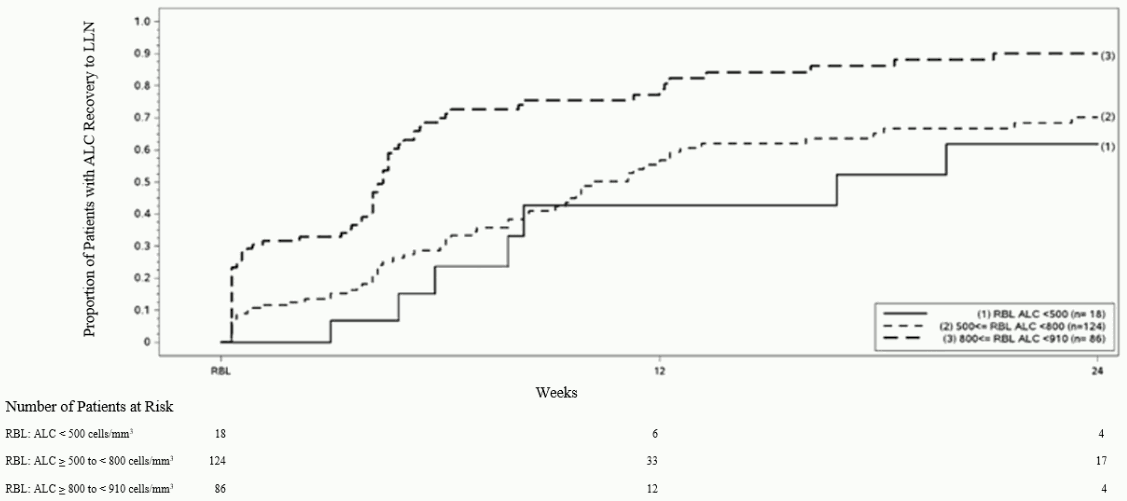
<bTable 1. Kaplan-Meier Method; Proportion of patients estimated to reach LLN, mild lymphopenia at the recovery baseline (RBL), excluding patients with prolonged severe lymphopenia:
| Number of patients with mild lymphopeniaa at risk | Baseline N=86 | Week 12 N=12 | Week 24 N=4 |
|---|---|---|---|
| Proportion reaching LLN (95% CI) | 0.81 (0.71, 0.89) | 0.90 (0.81, 0.96) |
a Patients with ALC <910 and ≥800 cells/mm³ at RBL, excluding patients with prolonged severe lymphopenia.
Table 2. Kaplan-Meier Method; Proportion of patients estimated to reach LLN, moderate lymphopenia at the recovery baseline (RBL), excluding patients with prolonged severe lymphopenia:
| Number of patients with moderate lymphopeniaa at risk | Baseline N=124 | Week 12 N=33 | Week 24 N=17 |
|---|---|---|---|
| Proportion reaching LLN (95% CI) | 0.57 (0.46, 0.67) | 0.70 (0.60, 0.80) |
a Patients with ALC <800 and ≥500 cells/mm³ at RBL, excluding patients with prolonged severe lymphopenia.
Table 3. Kaplan-Meier Method; Proportion of patients estimated to reach LLN, severe lymphopenia at the recovery baseline (RBL), excluding patients with prolonged severe lymphopenia:
| Number of patients with severe lymphopeniaa at risk | Baseline N=18 | Week 12 N=6 | Week 24 N=4 |
|---|---|---|---|
| Proportion reaching LLN (95% CI) | 0.43 (0.20, 0.75) | 0.62 (0.35, 0.88) |
a Patients with ALC <500 cells/mm³ at RBL, excluding patients with prolonged severe lymphopenia.
Clinical efficacy and safety
Two, 2 year, randomised, double-blind, placebo-controlled studies (DEFINE with 1,234 patients and CONFIRM with 1,417 patients) of patients with relapsing-remitting multiple sclerosis (RRMS) were performed. Patients with progressive forms of MS were not included in these studies.
Efficacy (see table below) and safety were demonstrated in patients with Expanded Disability Status Scale (EDSS) scores ranging from 0 to 5 inclusive, who had experienced at least 1 relapse during the year prior to randomisation, or, in the 6 weeks before randomisation had a brain Magnetic Resonance Imaging (MRI) demonstrating at least one gadolinium-enhancing (Gd+) lesion. Study CONFIRM contained a rater-blinded (i.e. study physician/investigator assessing the response to study treatment was blinded) reference comparator of glatiramer acetate.
In DEFINE, patients had the following median baseline characteristics: age 39 years, disease duration 7.0 years, EDSS score 2.0. In addition, 16% of patients had an EDSS score >3.5, 28% had ≥2 relapses in the prior year and 42% had previously received other approved MS treatments. In the MRI cohort 36% of patients entering the study had Gd+ lesions at baseline (mean number of Gd+ lesions 1.4).
In CONFIRM, patients had the following median baseline characteristics: age 37 years, disease duration 6.0 years, EDSS score 2.5. In addition, 17% of patients had an EDSS score >3.5, 32% had ≥2 relapses in the prior year and 30% had previously received other approved MS treatments. In the MRI cohort 45% of patients entering the study had Gd+ lesions at baseline (mean number of Gd+ lesions 2.4).
Compared to placebo, patients treated with Tecfidera had a clinically meaningful and statistically significant reduction on the primary endpoint in study DEFINE, proportion of patients relapsed at 2 years; and the primary endpoint in study CONFIRM, annualised relapse rate (ARR) at 2 years.
The ARR for glatiramer acetate and placebo was 0.286 and 0.401 respectively in study CONFIRM, corresponding to a reduction of 29% (p=0.013), which is consistent with approved prescribing information.
| DEFINE | CONFIRM | ||||
|---|---|---|---|---|---|
| Placebo | Tecfidera 240 mg Placebo | Placebo | Tecfidera 240 mg twice a day | Glatiramer acetate | |
| Clinical Endpointsa | |||||
| No. patients | 408 | 410 | 363 | 359 | 350 |
| Annualised relapse rate | 0.364 | 0.172*** | 0.401 | 0.224*** | 0.286* |
| Rate ratio (95% CI) | 0.47 (0.37, 0.61) | 0.56 (0.42, 0.74) | 0.71 (0.55, 0.93) | ||
| Proportion relapsed | 0.461 | 0.270*** | 0.410 | 0.291** | 0.321** |
| Hazard ratio (95% CI) | 0.51 (0.40, 0.66) | 0.66 (0.51, 0.86) | 0.71 (0.55, 0.92) | ||
| Proportion with 12-week confirmed disability progression | 0.271 | 0.164** | 0.169 | 0.128# | 0.156# |
| Hazard ratio (95% CI) | 0.62 (0.44, 0.87) | 0.79 (0.52, 1.19) | 0.93 (0.63, 1.37) | ||
| Proportion with 24 week confirmed disability progression | 0.169 | 0.128# | 0.125 | 0.078# | 0.108# |
| Hazard ratio (95% CI) | 0.77 (0.52, 1.14) | 0.62 (0.37, 1.03) | 0.87 (0.55, 1.38) | ||
| MRI Endpointsb | |||||
| No. patients | 165 | 152 | 144 | 147 | 161 |
| Mean (median) number of new or newly enlarging T2 lesions over 2 years | 16.5 (7.0) | 3.2 (1.0)*** | 19.9 (11.0) | 5.7 (2.0)*** | 9.6 (3.0)*** |
| Lesion mean ratio (95% CI) | 0.15 (0.10, 0.23) | 0.29 (0.21, 0.41) | 0.46 (0.33, 0.63) | ||
| Mean (median) number of Gd lesions at 2 years | 1.8 (0) | 0.1 (0)*** | 2.0 (0.0) | 0.5 (0.0)*** | 0.7 (0.0)** |
| Odds ratio (95% CI) | 0.10 (0.05, 0.22) | 0.26 (0.15, 0.46) | 0.39 (0.24, 0.65) | ||
| Mean (median) number of new T1 hypointense lesions over 2 years | 5.7 (2.0) | 2.0 (1.0)*** | 8.1 (4.0) | 3.8 (1.0)*** | 4.5 (2.0)** |
| Lesion mean ratio (95% CI) | 0.28 (0.20, 0.39) | 0.43 (0.30, 0.61) | 0.59 (0.42, 0.82) | ||
a All analyses of clinical endpoints were intent-to-treat
b MRI analysis used MRI cohort
* P-value <0.05
** P-value <0.01
*** P-value <0.0001
# not statistically significant
An open non-controlled 8-year extension study (ENDORSE) enrolled 1,736 eligible RRMS patients from the pivotal studies (DEFINE and CONFIRM). The primary objective of the study was to assess the long-term safety of Tecfidera in patients with RRMS. Of the 1,736 patients, approximately half (909, 52%) were treated for 6 years or longer. 501 patients were continuously treated with Tecfidera 240 mg twice daily across all 3 studies and 249 patients who were previously treated with placebo in studies DEFINE and CONFIRM received treatment 240 mg twice daily in study ENDORSE. Patients who received treatment twice daily continuously were treated for up to 12 years.
During study ENDORSE, more than half of all patients treated with Tecfidera 240 mg twice daily did not have a relapse. For patients continuously treated twice daily across all 3 studies, the adjusted ARR was 0.187 (95% CI: 0.156, 0.224) in studies DEFINE and CONFIRM and 0.141 (95% CI: 0.119, 0.167) in study ENDORSE. For patients previously treated with placebo, the adjusted ARR decreased from 0.330 (95% CI: 0.266, 0.408) in studies DEFINE and CONFIRM to 0.149 (95% CI: 0.116, 0.190) in study ENDORSE.
In study ENDORSE, the majority of patients (>75%) did not have confirmed disability progression (measured as 6-month sustained disability progression). Pooled results from the three studies demonstrated Tecfidera treated patients had consistent and low rates of confirmed disability progression with slight increase in mean EDSS scores across ENDORSE. MRI assessments (up to year 6, including 752 patients who had previously been included in the MRI cohort of studies DEFINE and CONFIRM showed that the majority of patients (approximately 90%) had no Gd-enhancing lesions. Over the 6 years, the annual adjusted mean number of new or newly enlarging T2 and new T1 lesions remained low.
Efficacy in patients with high disease activity
In studies DEFINE and CONFIRM, consistent treatment effect on relapses in a subgroup of patients with high disease activity was observed, whilst the effect on time to 3-month sustained disability progression was not clearly established. Due to the design of the studies, high disease activity was defined as follows:
- Patients with 2 or more relapses in one year, and with one or more Gd-enhancing lesions on brain MRI (n=42 in DEFINE; n=51 in CONFIRM) or,
- Patients who have failed to respond to a full and adequate course (at least one year of treatment) of beta-interferon, having had at least 1 relapse in the previous year while on therapy, and at least 9 T2-hyperintense lesions in cranial MRI or at least 1 Gd-enhancing lesion, or patients having an unchanged or increased relapse rate in the prior year as compared to the previous 2 years (n=177 in DEFINE; n=141 in CONFIRM).
Paediatric population
Dimethyl fumarate was evaluated in a prospective open-label, uncontrolled study in 22 paediatric patients with RRMS aged 13 to 17 years (4 patients aged ≤ 14 years). Patients received Dimethyl fumarate 120 mg twice a day for 7 days followed by 240 mg twice a day for 24 weeks. The median number of new or newly enlarging T2 hyperintense lesions changed from 2 in the 8-week pretreatment evaluation period to 0 in the final 8 weeks of the treatment period (median change 2, n = 16). Patients subsequently entered an extension study for a further 96 weeks. Among the 10 patients with MRI data between weeks 64 and week 72 of the extension study, the median number of subjects with new or newly enlarging T2 hyperintense lesions was 0 (range 0,2). Over the full treatment period (120-week), ARR was 0.2 representing an 84.5% relative reduction in relapses (n = 20; 95% CI [66.8, 92.8], p< 0.0001), when compared to the year prior to treatment initiation. These data should be considered cautiously regarding limitations of the study design (no control arm, pre versus post-dose comparison) (see section 4.2).
5.2. Pharmacokinetic properties
Orally administered dimethyl fumarate undergoes rapid presystemic hydrolysis by esterases and is converted to its primary metabolite, monomethyl fumarate, which is also active. Dimethyl fumarate is not quantifiable in plasma following oral administration of dimethyl fumarate. Therefore, all pharmacokinetic analyses related to dimethyl fumarate were performed with plasma monomethyl fumarate concentrations. Pharmacokinetic data were obtained in subjects with multiple sclerosis and healthy volunteers.
Absorption
The Tmax of monomethyl fumarate is 2 to 2.5 hours. As Dimethyl fumarate Mylan gastro-resistant hard capsules contain enteric coated pellets, which are protected by an enteric coating, absorption does not commence until they leave the stomach (generally less than 1 hour). Following 240 mg twice a day administered with food, the median peak (Cmax) was 1.72 mg/l and overall area under the curve (AUC) exposure was 8.02 h.mg/l in subjects with multiple sclerosis. Overall, Cmax and AUC increased approximately dose-proportionally in the dose range studied (120 mg to 360 mg). In subjects with multiple sclerosis, two 240 mg doses were administered 4 hours apart as part of a three times a day dosing regimen. This resulted in a minimal accumulation of exposure yielding an increase in the median Cmax of 12% compared to the twice daily dosing (1.72 mg/l for twice daily compared to 1.93 mg/l for three times daily) with no safety implications.
Food does not have a clinically significant effect on exposure of dimethyl fumarate. However, dimethyl fumarate should be taken with food due to improved tolerability with respect to flushing or gastrointestinal adverse events (see section 4.2).
Distribution
The apparent volume of distribution following oral administration of 240 mg dimethyl fumarate varies between 60 L and 90 L. Human plasma protein binding of monomethyl fumarate generally ranges between 27% and 40%.
Biotransformation
In humans, dimethyl fumarate is extensively metabolised with less than 0.1% of the dose excreted as unchanged dimethyl fumarate in urine. It is initially metabolised by esterases, which are ubiquitous in the gastrointestinal tract, blood and tissues, before it reaches the systemic circulation. Further metabolism occurs through the tricarboxylic acid cycle, with no involvement of the cytochrome P450 (CYP) system. A single 240 mg 14C-dimethyl fumarate dose study identified glucose as the predominant metabolite in human plasma. Other circulating metabolites included fumaric acid, citric acid and monomethyl fumarate. The downstream metabolism of fumaric acid occurs through the tricarboxylic acid cycle, with exhalation of CO2 serving as a primary route of elimination.
Elimination
Exhalation of CO2 is the primary route of dimethyl fumarate elimination accounting for 60% of the dose. Renal and faecal elimination are secondary routes of elimination, accounting for 15.5% and 0.9% of the dose respectively.
The terminal half-life of monomethyl fumarate is short (approximately 1 hour) and no circulating monomethyl fumarate is present at 24 hours in the majority of individuals. Accumulation of parent drug or monomethyl fumarate does not occur with multiple doses of dimethyl fumarate at the therapeutic regimen.
Linearity
Dimethyl fumarate exposure increases in an approximately dose proportional manner with single and multiple doses in the 120 mg to 360 mg dose range studied.
Pharmacokinetics in special patient groups
Based on the results of Analysis of Variance (ANOVA), body weight is the main covariate of exposure (by Cmax and AUC) in RRMS subjects, but did not affect safety and efficacy measures evaluated in the clinical studies.
Gender and age did not have a clinically significant impact on the pharmacokinetics of dimethyl fumarate. The pharmacokinetics in patients aged 65 and over has not been studied.
Paediatric population
The pharmacokinetic profile of 240 mg dimethyl fumarate twice a day was evaluated in a small, openlabel, uncontrolled study in patients with RRMS aged 13 to 17 years (n = 21). The pharmacokinetics of dimethyl fumarate in these adolescent patients was consistent with that previously observed in adult patients (Cmax: 2.00±1.29 mg/l; AUC0-12hr: 3.62±1.16 hmg/l, which corresponds to an overall daily AUC of 7.24 hmg/l).
Renal impairment
Since the renal pathway is a secondary route of elimination for dimethyl fumarate accounting for less than 16% of the dose administered, evaluation of pharmacokinetics in individuals with renal impairment was not conducted.
Hepatic impairment
As dimethyl fumarate and monomethyl fumarate are metabolised by esterases, without the involvement of the CYP450 system, evaluation of phamacokinetics in individuals with hepatic impairment was not conducted.
5.3. Preclinical safety data
The adverse reactions described in the Toxicology and Reproduction toxicity sections below were not observed in clinical studies, but were seen in animals at exposure levels similar to clinical exposure levels.
Mutagenesis
Dimethyl fumarate and mono-methylfumarate were negative in a battery of in vitro assays (Ames, chromosomal aberration in mammalian cells). Dimethyl fumarate was negative in the in vivo micronucleus assay in the rat.
Carcinogenesis
Carcinogenicity studies of dimethyl fumarate were conducted for up to 2 years in mice and rats. Dimethyl fumarate was administered orally at doses of 25, 75, 200 and 400 mg/kg/day in mice, and at doses of 25, 50, 100, and 150 mg/kg/day in rats. In mice, the incidence of renal tubular carcinoma was increased at 75 mg/kg/day, at equivalent exposure (AUC) to the recommended human dose. In rats, the incidence of renal tubular carcinoma was increased at 100 mg/kg/day, approximately 2 times higher exposure than the recommended human dose. The relevance of these findings to human risk is unknown.
The incidence of squamous cell papilloma and carcinoma in the nonglandular stomach (forestomach) was increased at equivalent exposure to the recommended human dose in mice and below exposure to the recommended human dose in rats (based on AUC). The forestomach in rodents does not have a human counterpart.
Toxicology
Nonclinical studies in rodent, rabbits, and monkeys were conducted with a dimethyl fumarate suspension (dimethyl fumarate in 0.8% hydroxypropyl methylcellulose) administered by oral gavage. The chronic dog study was conducted with oral administration of the dimethyl fumarate capsule.
Kidney changes were observed after repeated oral administration of dimethyl fumarate in mice, rats, dogs, and monkeys. Renal tubule epithelial regeneration, suggestive of injury, was observed in all species. Renal tubular hyperplasia was observed in rats with life time dosing (2-year study). In dogs that received daily oral doses of dimethyl fumarate for 11 months, the margin calculated for cortical atrophy was observed at 3 times the recommended dose based on AUC. In monkeys that received daily oral doses of dimethyl fumarate for 12 months, single cell necrosis was observed at 2 times the recommended dose based on AUC. Interstitial fibrosis and cortical atrophy were observed at 6 times the recommended dose based on AUC. The relevance of these findings to humans is not known.
In the testes, degeneration of the seminiferous epithelium was seen in rats and dogs. The findings were observed at approximately the recommended dose in rats and 3 times the recommended dose in dogs (AUC basis). The relevance of these findings to humans is not known.
Findings in the forestomach of mice and rats consisted of squamous epithelial hyperplasia and hyperkeratosis; inflammation; and squamous cell papilloma and carcinoma in studies of 3 months or longer in duration. The forestomach of mice and rats does not have a human counterpart.
Reproduction toxicity
Oral administration of dimethyl fumarate to male rats at 75, 250, and 375 mg/kg/day prior to and during mating had no effects on male fertility up to the highest dose tested (at least 2 times the recommended dose on an AUC basis). Oral administration of dimethyl fumarate to female rats at 25, 100, and 250 mg/kg/day prior to and during mating, and continuing to Day 7 of gestation, induced reduction in the number of estrous stages per 14 days and increased the number of animals with prolonged diestrus at the highest dose tested (11 times the recommended dose on an AUC basis). However, these changes did not affect fertility or the number of viable fetuses produced.
Dimethyl fumarate has been shown to cross the placental membrane into fetal blood in rats and rabbits, with ratios of fetal to maternal plasma concentrations of 0.48 to 0.64 and 0.1 respectively. No malformations were observed at any dose of dimethyl fumarate in rats or rabbits. Administration of dimethyl fumarate at oral doses of 25, 100, and 250 mg/kg/day to pregnant rats during the period of organogenesis resulted in maternal adverse effects at 4 times the recommended dose on an AUC basis, and low fetal weight and delayed ossification (metatarsals and hindlimb phalanges) at 11 times the recommended dose on an AUC basis. The lower fetal weight and delayed ossification were considered secondary to maternal toxicity (reduced body weight and food consumption).
Oral administration of dimethyl fumarate at 25, 75, and 150 mg/kg/day to pregnant rabbits during organogenesis had no effect on embryo-fetal development and resulted in reduced maternal body weight at 7 times the recommended dose and increased abortion at 16 times the recommended dose, on an AUC basis.
Oral administration of dimethyl fumarate at 25, 100, and 250 mg/kg/day to rats during pregnancy and lactation resulted in lower body weights in the F1 offspring, and delays in sexual maturation in F1 males at 11 times the recommended dose on an AUC basis. There were no effects on fertility in the F1 offspring. The lower offspring body weight was considered secondary to maternal toxicity.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.