Source: Health Products Regulatory Authority (ZA) Revision Year: 2022 Publisher: Austell Pharmaceuticals (Pty) Ltd, 1 Sherborne Road, Parktown, JOHANNESBURG, 2193, South Africa Tel: 0860287835 Manufactured by Lupin Ltd, imported and distributed by Austell Pharmaceuticals (Pty) Ltd. ...
WARNING
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues alone or in combination with other antiretrovirals (see section 4.4).
DOLTELAM is not indicated for the treatment of chronic hepatitis B virus (HBV) infection. The safety and efficacy of DOLTELAM has not been established in patients co infected with HBV and HIV. Severe acute exacerbations of Hepatitis B have been reported in patients who are co-infected with HBV and HIV and have discontinued the combination tablet.
Hepatic function should be monitored closely with both clinical and laboratory follow-up for at least several months in patients who discontinue DOLTELAM and are co-infected with HBV and HIV. If appropriate, initiation of anti-hepatitis B therapy may be warranted (see section 4.4).
Safety and efficacy of the individual active ingredients in various antiretroviral combination regimens with similar dosages as those contained in DOLTELAM have been established in clinical studies for the treatment of HIV patients. However, safety and efficacy of the fixed-medicine combination, as in DOLTELAM, for the treatment of HIV has not been established in clinical studies. The complete professional information of each of the other medicines used in combination should be consulted before initiation of therapy.
HBV antibody testing should be offered to all individuals before initiating lamivudine and tenofovir disoproxil-containing therapies (see below Patients with HIV and hepatitis B (HBV) or C virus (HCV) co-infections).
Combination antiretroviral therapy, including DOLTELAM, has been associated with metabolic abnormalities such as hypertriglyceridemia, hypercholesterolaemia, insulin resistance, hyperglycaemia and hyperlactataemia.
Redistribution/accumulation of body fat, including central obesity, dorsocervical fat enlargement (buffalo hump), peripheral wasting, facial wasting, breast enlargement, elevated serum lipid and glucose levels have been observed either separately or together in some patients receiving combination antiretroviral therapy. A higher risk of lipodystrophy has been associated with individual factors such as older age, and with medicine-related factors such as longer duration of antiretroviral treatment and associated metabolic disturbances. Clinical examination should include evaluation for physical signs of fat redistribution.
Consideration should be given to the measurement of serum lipids and blood glucose. Lipid disorders should be managed as clinically appropriate. Patients with evidence of lipodystrophy should also have a thorough cardiovascular risk assessment.
Immune Reconstitution Inflammatory Syndrome (IRIS) is an immunopathological response resulting from the rapid restoration of pathogen-specific immune responses to pre-existing antigens combined with immune dysregulation, which occurs shortly after starting combination AntiRetroviral Therapy (cART). Typically, such reactions present by paradoxical deterioration of opportunistic infections being treated or with unmasking of an asymptomatic opportunistic disease, often with an atypical inflammatory presentation. IRIS usually develops within the first three months of initiation of antiretroviral therapy (ART) and occurs more commonly in patients with low CD4 counts. Common examples of IRIS reactions to opportunistic diseases are tuberculosis, atypical mycobacterial infections, cytomegalovirus retinitis, Pneumocystis jirovecii, and cryptococcal meningitis.
Appropriate treatment of the opportunistic disease should be instituted or continued and ART continued. Inflammatory manifestations generally subside after a few weeks.
Severe cases may respond to glucocorticoids, but there is only limited evidence for this in patients with tuberculosis IRIS. Autoimmune disorders (such as Graves' disease, Guillain-Barre Syndrome, Polymyositis) have also been reported as IRIS reactions; however, the reported time to onset is more variable and these events can occur many months after initiation of treatment.
Raised liver enzymes, consistent with IRIS, occurred in some patients who also had hepatitis B or C infection at the start of dolutegravir therapy. Monitoring of liver function is recommended in patients with hepatitis B or C infection. Particular care should be taken in initiating or maintaining effective hepatitis B therapy when starting dolutegravir-based therapy in patients with hepatitis B.
Osteonecrosis has been reported particularly in patients with advanced HIV disease or following long-term combination cART. Their aetiology can be multifactorial and include corticosteroid use, excessive alcohol consumption, severe immunosuppression, and being overweight. Patients should be advised to speak to their health care provider if they have joint aches and pain, joint stiffness or difficulty in movement.
Patients receiving DOLTELAM may continue to develop opportunistic infections and other complications of HIV infection. Therefore, patients should remain under close clinical observation by health care providers experienced in the treatment of HIV associated diseases. Regular monitoring of viral load and CD4 counts needs to be done.
Patients should be advised that treatment with DOLTELAM, has not been proven to prevent the risk of transmission of HIV to others through sexual contact or blood contamination. Appropriate precautions should continue to be taken.
Lactic acidosis, usually associated with hepatic steatosis, including fatal cases, has been reported with the use of nucleoside analogues, such as in DOLTELAM. Early symptoms (symptomatic hyperlactataemia) include benign digestive symptoms (nausea, vomiting and abdominal pain), nonspecific malaise, loss of appetite, weight loss, respiratory symptoms (rapid and/or deep breathing) or neurological symptoms (including motor weakness). Lactic acidosis has a high mortality and may be associated with pancreatitis, liver failure or renal failure.
Lactic acidosis generally occurs after a few or several months of treatment. Treatment with nucleoside analogues should be discontinued in the setting of symptomatic hyperlactataemia and metabolic/lactic acidosis, progressive hepatomegaly, or rapidly elevating aminotransferase levels. Suspicious biochemical features include mild raised transaminases, raised lactate dehydrogenase (LDH) and/or creatine kinase.
In patients with suspicious symptoms or biochemistry, measure the venous lactate level (normal <2 mmol/litre) and respond as follows:
The above lactate values may not be applicable to paediatric patients.
Diagnosis of lactic acidosis is confirmed by demonstrating metabolic acidosis with an increased anion gap and raised lactate level. Therapy should be stopped in any acidotic patient with a raised lactate level.
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases have been reported with the use of DOLTELAM alone or in combination, in the treatment of HIV infection. Most cases were women. Caution should be exercised when administering DOLTELAM to patients with known risk factors for liver disease.
Treatment with DOLTELAM should be suspended in any patient who develops clinical or laboratory findings suggestive of lactic acidosis or hepatoxicity. Caution should be exercised when administering nucleoside analogues as contained in DOLTELAM to any patient (particularly obese women) with hepatomegaly, hepatitis or other known risk factors for liver disease and hepatic steatosis (including certain medicines and alcohol). Patients co-infected with Hepatitis C and treated with alpha interferon and ribavirin may constitute a special risk. Patients at increased risk should be followed closely. However, cases have also been reported in patients with no known risk factors.
There are no study results demonstrating the effect of DOLTELAM on clinical progression of HIV-1.
Nucleoside and nucleotide analogues as contained in DOLTELAM can cause a variable degree of mitochondrial damage in vitro and in vivo. There have been reports of mitochondrial dysfunction in HIV-negative infants exposed in utero and/or postnatally to nucleoside analogues; these have predominantly concerned treatment with regimens containing zidovudine. The main adverse events are haematological (anaemia, neutropenia) and metabolic disorders (hyperlactataemia, hyperlipidaemia). These events are often transitory. Some late-onset neurological disorders have been reported (hypertonia, convulsion, abnormal behaviour). Whether the neurological disorders are transient or permanent is currently unknown. Any child exposed in utero to nucleoside and nucleotide analogues, even HIV-negative children, should have clinical and laboratory follow-up and should be fully investigated for possible mitochondrial dysfunction in case of relevant signs or symptoms.
Pancreatitis has been observed in some patients receiving lamivudine, as in DOLTELAM. It is unclear whether this is due to lamivudine or to underlying HIV disease. Pancreatitis must be considered whenever a patient develops abdominal pain, nausea, vomiting or elevated biochemical markers. Treatment with DOLTELAM should be stopped immediately if clinical signs, symptoms or laboratory abnormalities suggestive of pancreatitis occur (see section 4.8).
The terminal half-life of DOLTELAM is increased in patients with moderate to severe renal impairment due to decreased clearance (see section 4.3).
DOLTELAM is a combination medicine and the dose of the individual components cannot be altered. Since DOLTELAM is primarily eliminated by the kidneys, co-administration of DOLTELAM with medicines that reduce renal function or compete for active tubular secretion may increase serum concentrations of DOLTELAM and/or increase the concentrations of other renally eliminated medicines. Some examples include, but are not limited to adefovir dipivoxil, cidofovir, aciclovir, valaciclovir, ganciclovir and valganciclovir.
DOLTELAM is not recommended for patients with creatinine clearance < 80 mL/min, or patients requiring haemodialysis. Renal failure, renal impairment, elevated creatinine, hypophosphatemia and proximal tubulopathy (including Fanconi syndrome) have been reported with the use of tenofovir disoproxil in clinical practice (see section 4.8). Careful monitoring of renal function (serum creatinine and serum phosphate) is therefore recommended before therapy commences.
Renal safety with tenofovir has only been studied to a very limited degree in adult patients with impaired renal function (creatinine clearance < 80mL/min).
Renal function (creatinine clearance and serum phosphate) assessment in all patients, prior to initiating therapy with tenofovir disoproxil fumarate, with monitoring every four weeks during the first year of treatment and every three months thereafter, is recommended. In patients at risk for renal impairment, including patients who have previously experienced renal events while receiving adefovir dipivoxil, consideration should be given to more frequent monitoring of renal function.
DOLTELAM should be avoided with concurrent or recent use of a nephrotoxic medicine (e.g. highdose or multiple non-steroidal anti-inflammatory medicines, aminoglycosides, amphotericin B, foscarnet, ganciclovir, pentamidine, vancomycin, cidofovir, interleukin-2). If concomitant use of DOLTELAM and nephrotoxic medicine is unavoidable, patients at risk of, or with a history of, renal dysfunction and patients receiving concomitant nephrotoxic substances should be carefully monitored for changes in serum creatinine and phosphorous (see section 4.5).
Tenofovir disoproxil fumarate has not been clinically evaluated in patients receiving medicines which are secreted by the same renal pathway, including the transport proteins human organic anion transporter (hOAT) 1 and 3 or MRP 4 (e.g. cidofovir, a known nephrotoxic medicine). These renal transport proteins may be responsible for tubular secretion and in part, renal elimination of tenofovir and cidofovir. Consequently, the pharmacokinetics of these medicines, which are secreted by the same renal pathway including transport proteins hOAT 1 and 3 or MRP 4, might be modified if they are co-administered. Unless clearly necessary, concomitant use of these medicines, which are secreted by the same renal pathway, is not recommended, but if such use is unavoidable, renal function should be monitored weekly (see section 4.5).
DOLTELAM should be avoided in antiretroviral experienced patients with HIV-1 harbouring the K65R mutation.
Decreases in bone mineral density of spine and changes in bone biomarkers from baseline are significantly greater with tenofovir disoproxil fumarate, as contained in DOLTELAM. Decreases in bone mineral density of the hip is significantly greater. Clinically relevant bone fractures have been reported. If bone abnormalities are suspected then appropriate consultation should be obtained.
Bone monitoring should be considered for HIV infected patients who have a history of pathologic bone fracture or are at risk of osteopenia.
DOLTELAM may cause a reduction in bone mineral density. The effects of tenofovir disoproxil fumarate-associated changes in bone mineral density on long-term bone health and future fracture risk are currently unknown.
Bone abnormalities (infrequently contributing to fractures) may be associated with proximal renal tubulopathy (see section 4.8). If bone abnormalities are suspected, then appropriate consultation should be obtained. Bone monitoring should be considered for HIV infected patients who have a history of pathologic bone fracture or are at risk for osteopenia. Although the effect of supplementation with calcium and vitamin D was not studied, such supplementation may be beneficial for all patients.
Use of DOLTELAM can result in hepatomegaly due to non-alcoholic fatty liver disease (hepatic steatosis).
The safety and efficacy of DOLTELAM has not been established in patients with significant underlying liver disorders. Patients with pre-existing liver dysfunction including chronic active hepatitis, have an increased frequency of liver function abnormalities during combination antiretroviral therapy and should be monitored according to standard practice. If there is evidence of worsening liver disease in such patients, interruption or discontinuation of treatment must be considered.
DOLTELAM is not indicated for the treatment of chronic HBV infection. The safety and efficacy of DOLTELAM has not been established for the treatment of patients co-infected with HBV and HIV.
Patients with chronic hepatitis B or C and treated with combination antiretroviral therapy are at an increased risk of severe and potentially fatal hepatic adverse reactions. Healthcare providers should refer to current HIV treatment guidelines for the optimal management of HIV infection in patients co-infected with hepatitis B virus (HBV). In case of concomitant antiviral therapy for hepatitis B or C, the relevant product information for these medicines must be referred to.
Spontaneous exacerbations in chronic hepatitis B are relatively common and are characterised by transient increases in serum ALT. After initiating antiviral therapy, serum ALT may increase in some patients. In patients with compensated liver disease, these increases in serum ALT are generally not accompanied by an increase in serum bilirubin concentrations or hepatic decompensation. Patients with cirrhosis may be at a higher risk for hepatic decompensation following hepatitis exacerbation, and therefore should be monitored closely during therapy.
Acute exacerbations of hepatitis have been reported in patients after the discontinuation of hepatitis B therapy. Post-treatment exacerbations are usually associated with rising HBV DNA, and the majority appears to be self-limited. However, severe exacerbations, including fatalities, have been reported. Hepatic function should be monitored at repeated intervals with both clinical and laboratory follow-up for at least 6 months after discontinuation of hepatitis B therapy. If appropriate, resumption of hepatitis B therapy may be warranted. In patients with advanced liver disease or cirrhosis, treatment discontinuation is not recommended since post-treatment exacerbations of hepatitis may lead to hepatic decompensation. Liver flares are especially serious, and sometimes fatal in patients with decompensated liver disease.
Hypersensitivity reactions reported with integrase inhibitors, including dolutegravir as in DOLTELAM, and were characterised by rash, constitutional findings, and sometimes, organ dysfunction, including severe liver reactions. Dolutegravir and other suspect medicine should be discontinued immediately if hypersensitivity reactions develop (including severe rash or rash accompanied by raised liver enzymes, fever, general malaise, fatigue, muscle or joint aches, blisters, oral lesions, conjunctivitis, facial oedema, eosinophilia, and angioedema). Clinical status including liver aminotransferases and bilirubin should be monitored and appropriate therapy initiated. Delay in stopping treatment with DOLTELAM or other suspect medicine after the onset of hypersensitivity may result in a lifethreatening reaction.
The decision to use dolutegravir in the presence of HIV-1 resistance to integrase inhibitors should take into account that it is considerably less active against viral strains with Q148 with two or more secondary mutations from G140A/C/S, E138A/K/T, L74I. Dolutegravir’s contribution to efficacy is uncertain when it is used to treat HIV-1 with this type of resistance to integrase inhibitors.
Caution should be given to co-administering medicines (prescription and non-prescription) that may change the exposure of dolutegravir or medicines that may have their exposure changed by dolutegravir (see sections 4.3 and 4.5).
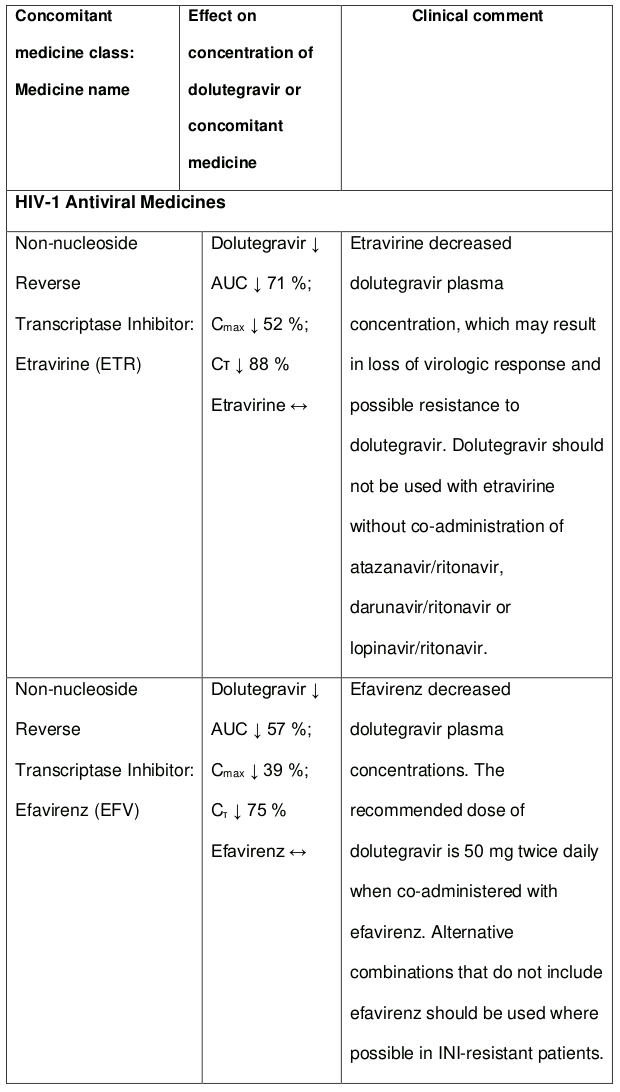
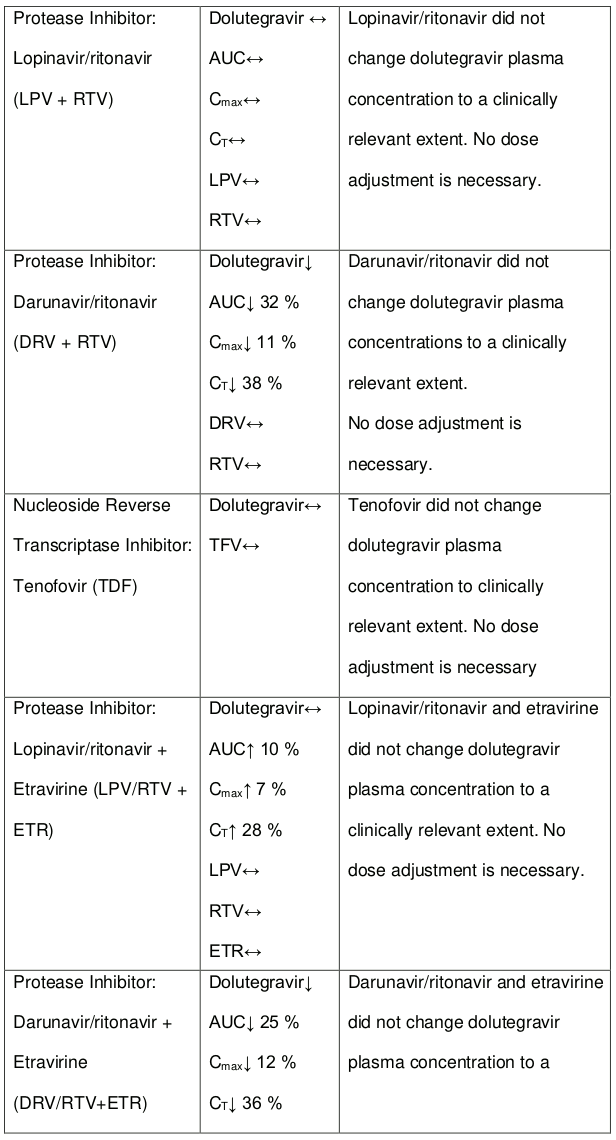
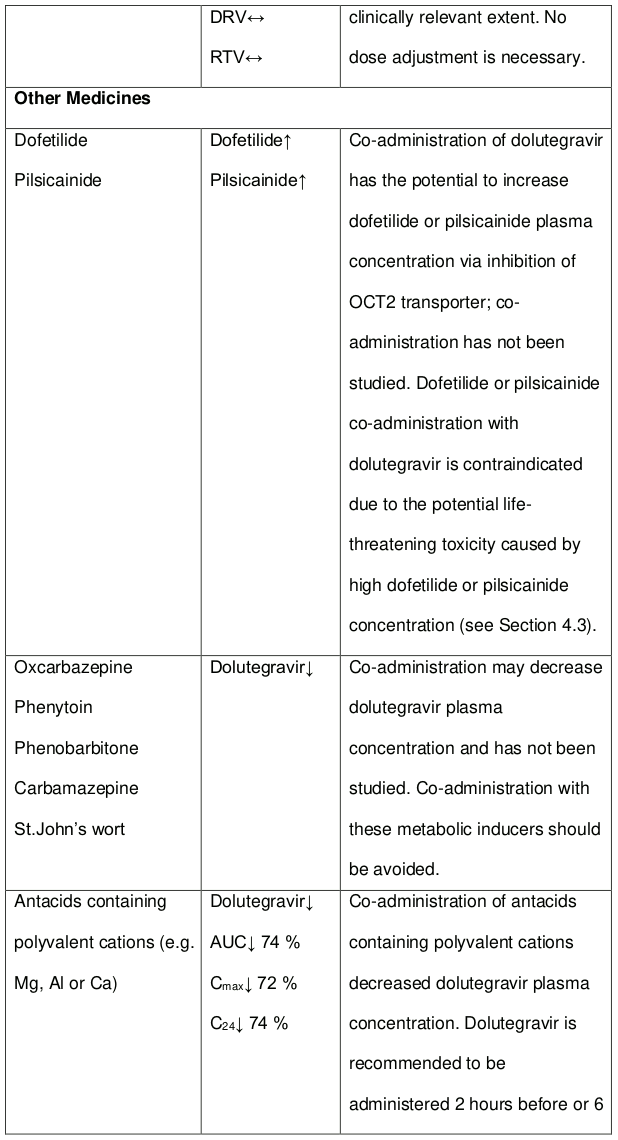
The co-administration of dolutegravir with etravirine (ETR) is not recommended unless the patient is also receiving concomitant atazanavir + ritonavir (ATV + RTV), lopinavir + ritonavir (LPV + RTV) or darunavir + ritonavir (DRV + RTV) (see section 4.5).
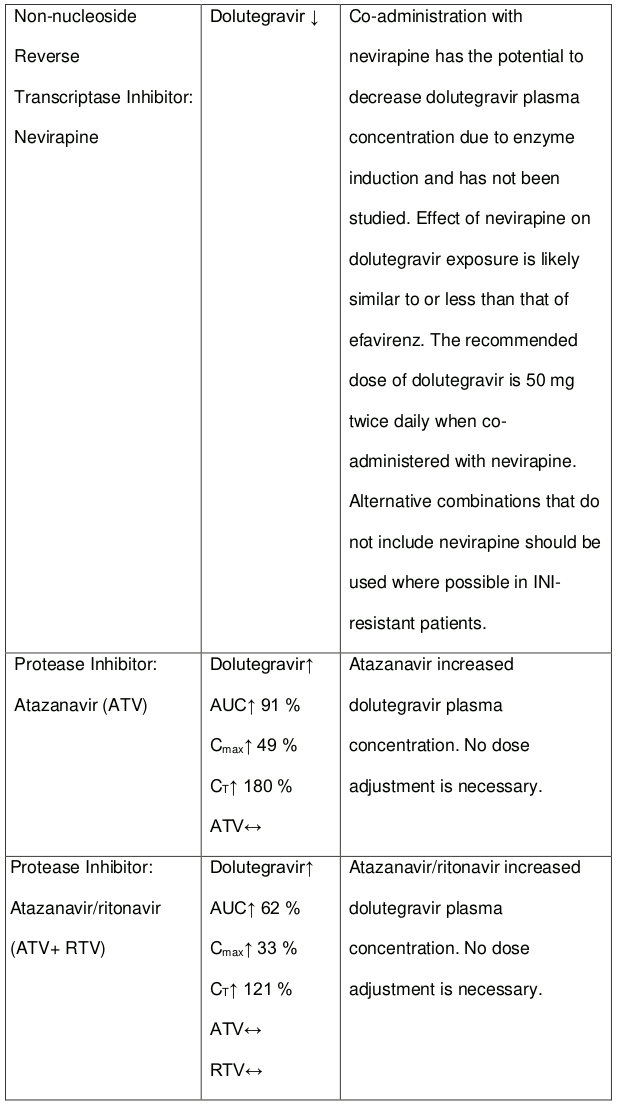
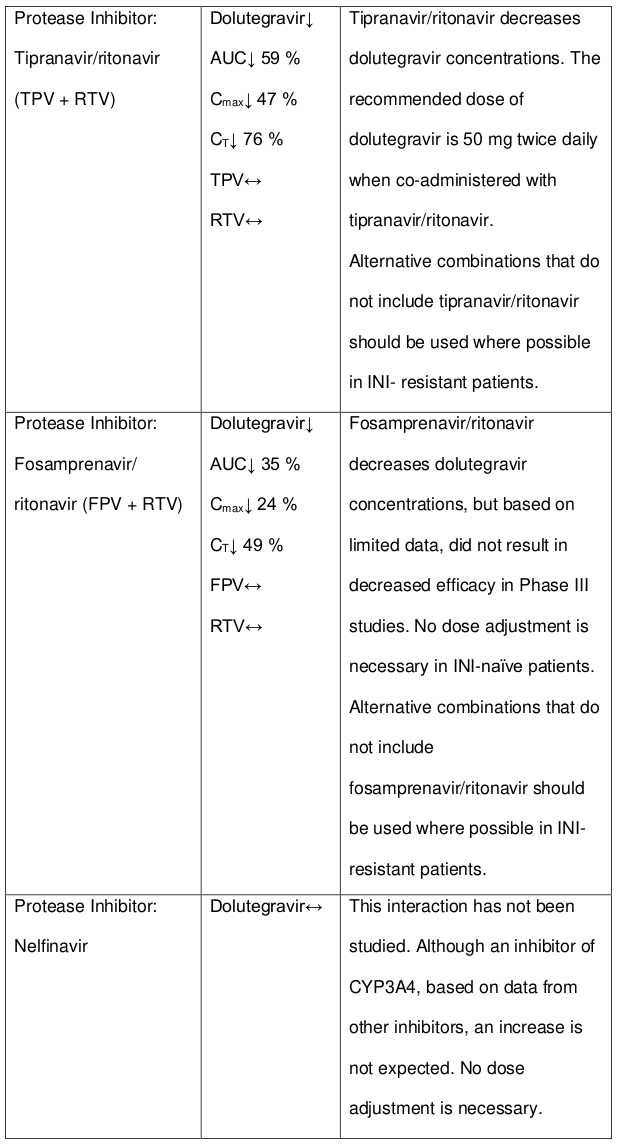
The recommended dose of dolutegravir is 50 mg twice daily when co-administered with efavirenz, nevirapine, tipranavir/ritonavir, or rifampicin (see section 4.5).
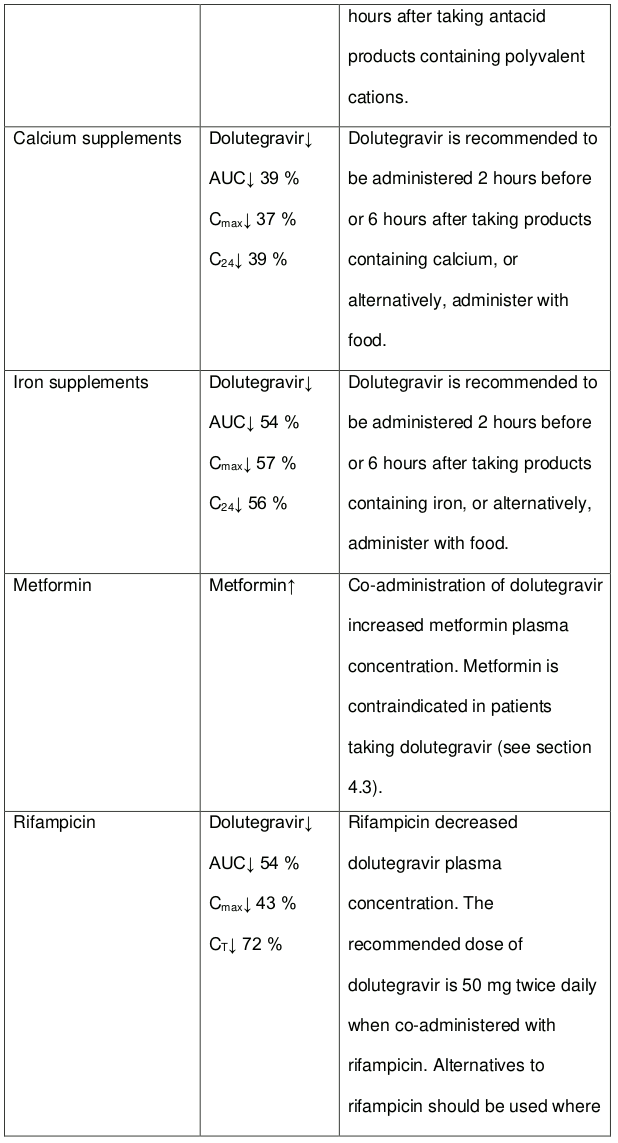
Dolutegravir should not be co-administered with polyvalent cation-containing antacids. Dolutegravir is recommended to be administered 2 hours before or 6 hours after these medicines (see section 4.5).
Metformin concentrations may be increased by dolutegravir. Metformin is contraindicated in patients taking dolutegravir (see section 4.3).
Safety and effectiveness in paediatric patients and patients <18 years of age have not been established.
Elderly patients are more likely to have decreased renal function; therefore, caution should be exercised when treating elderly patients with tenofovir disoproxil as in DOLTELAM. Clinical studies did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger patients.
DOLTELAM contains lactose and mannitol. Patients with rare hereditary problems of galactose intolerance, total lactase deficiency or glucose-galactose malabsorption should not take this medicine.
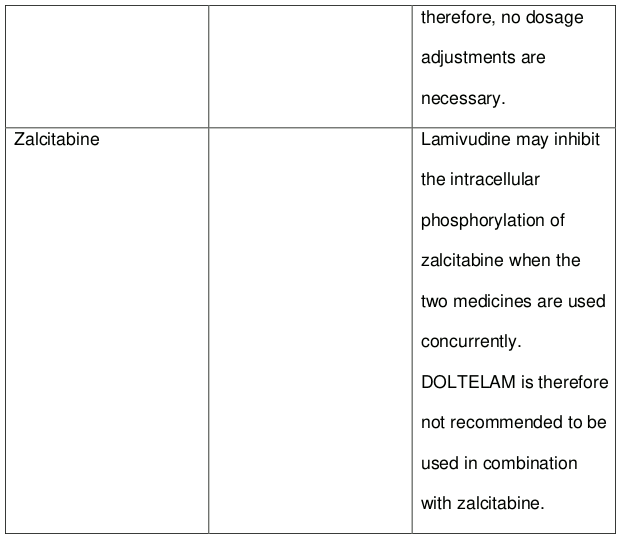
The likelihood of interactions is low due to the limited metabolism as plasma protein binding and almost complete renal clearance. Zidovudine plasma levels are not significantly altered when co-administered with lamivudine (as in DOLTELAM). Zidovudine has no effect on the pharmacokinetics of lamivudine. Lamivudine may inhibit the intracellular phosphorylation of zalcitabine when the two medicines are used concurrently. Lamivudine is therefore not recommended to be used in combination with zalcitabine.
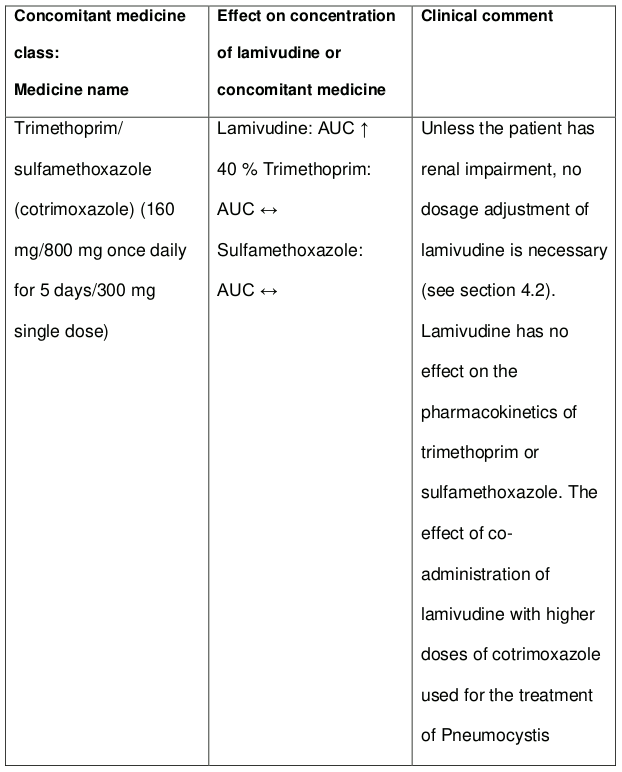
Administration of trimethoprim, a constituent of co-trimoxazole causes an increase in lamivudine plasma levels. However, unless the patient has renal impairment, no dosage adjustment of lamivudine is necessary. Lamivudine has no effect on the pharmacokinetics of co-trimoxazole. The possibility of interactions with other medicines administered concurrently should be considered, particularly when the main route is renal.
No medicine interaction studies have been conducted using DOLTELAM. As DOLTELAM contains tenofovir disoproxil fumarate and lamivudine, any interactions that have been identified with these individual medicines may occur with DOLTELAM. Important medicine interaction information for DOLTELAM is summarised in Tables 1, 2 and 3. The medicine interactions described are based on studies conducted with tenofovir disoproxil fumarate or lamivudine as individual medicines, or are potential medicine interactions. While the tables include potentially significant interactions, they are not all inclusive. Based on the results of in vitro experiments and the known elimination pathway of tenofovir, the potential for CYP450-mediated interactions involving tenofovir with other medicines is low.
An interaction with trimethoprim, a constituent of co-trimoxazole, causes a 40% increase in lamivudine exposure at therapeutic doses. This does not require dose adjustment unless the patient also has renal impairment. Administration of co-trimoxazole with the lamivudine/zidovudine combination in patients with renal impairment should be carefully assessed.
Tenofovir, as in DOLTELAM, is primarily excreted by the kidneys by a combination of glomerular filtration and active tubular secretion.
Co-administration of DOLTELAM with medicines that are eliminated by active tubular secretion may increase serum concentrations of either tenofovir or the co-administered medicines due to competition for this elimination pathway. Medicines that decrease renal function may also increase serum concentrations of tenofovir, as in DOLTELAM.
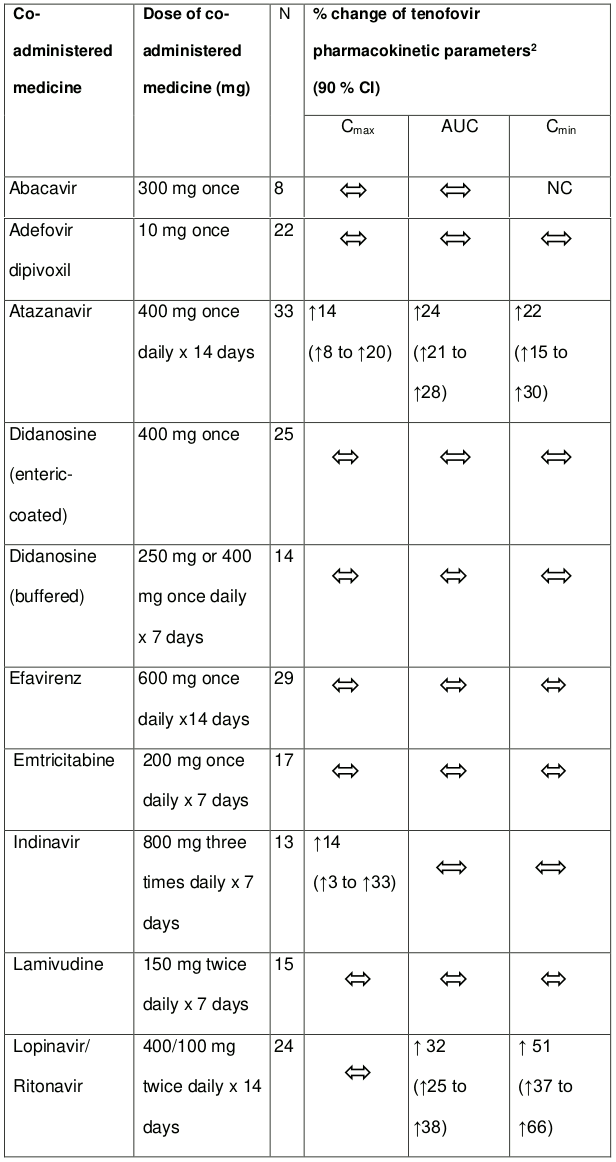
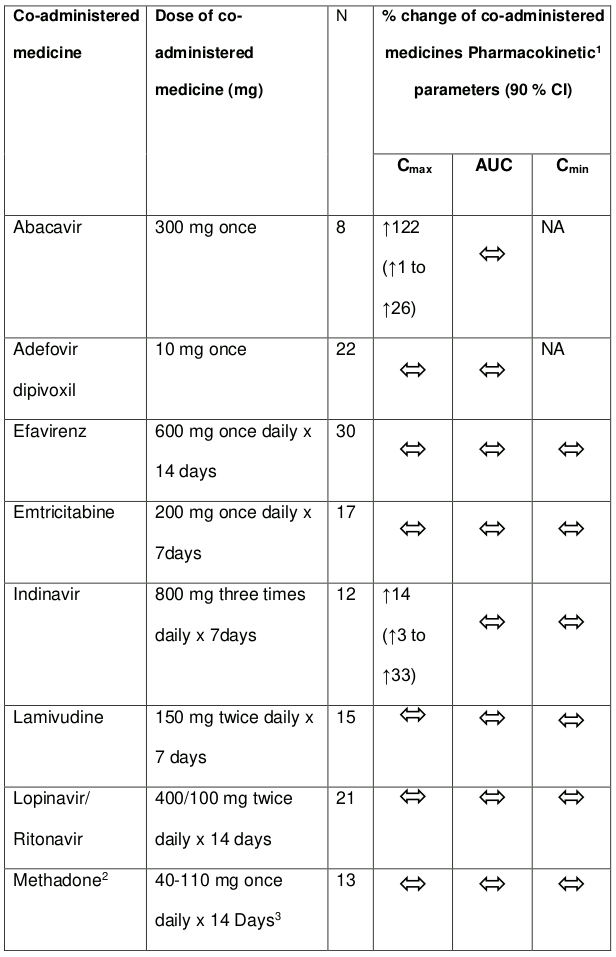
Tenofovir has been evaluated in healthy volunteers in combination with abacavir, adefovir dipivoxil, atazanavir, didanosine, efavirenz, indinavir, lamivudine, lopinavir/ritonavir, methadone, oral contraceptives and ribavirin. Tables 1 and 2 summarise pharmacokinetic effects of co-administered medicine on tenofovir pharmacokinetics and effects of tenofovir on the pharmacokinetics of coadministered medicine.
When administered with multiple doses of tenofovir, the Cmax and AUC of didanosine 400 mg increased significantly. The mechanism of this interaction is unknown. When didanosine 250 mg enteric-coated capsules were administered with tenofovir, systemic exposures to didanosine were similar to those seen with the 400 mg enteric-coated capsules alone under fasted conditions.
Table 1. Medicine interactions: Changes in pharmacokinetic parameters for Tenofovir1 in the presence of co-administered medicines:
1 Patients received as tenofovir disoproxil fumarate 300 mg once daily.
2 Increase =↑; Decrease =↓; No Effect = ; NC =Not calculated
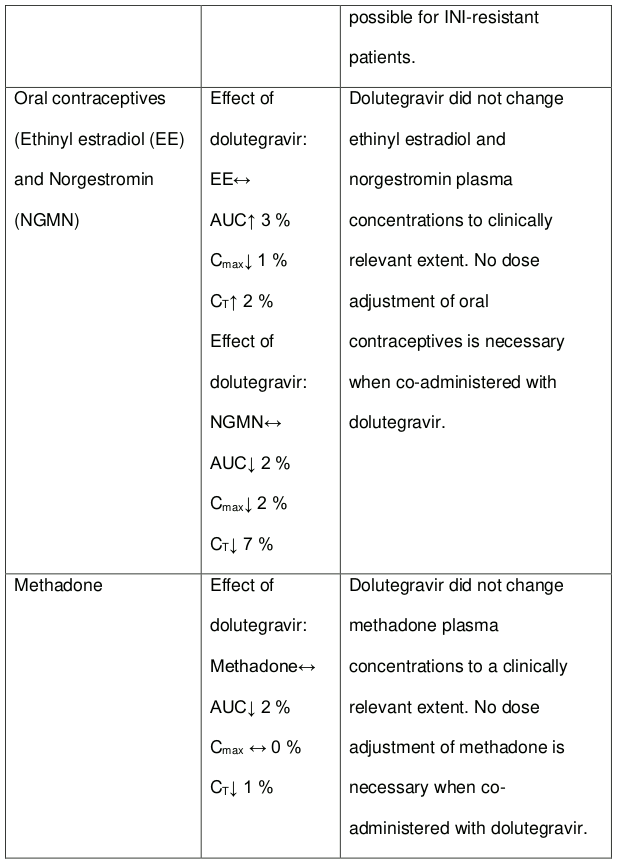
Following multiple dosing to HIV-negative patients receiving either chronic methadone maintenance therapy, oral contraceptives, or single doses of ribavirin, steady-state tenofovir pharmacokinetics were similar to those observed in previous studies, indicating a lack of clinically significant medicine interactions between these medicines and tenofovir disoproxil fumarate.
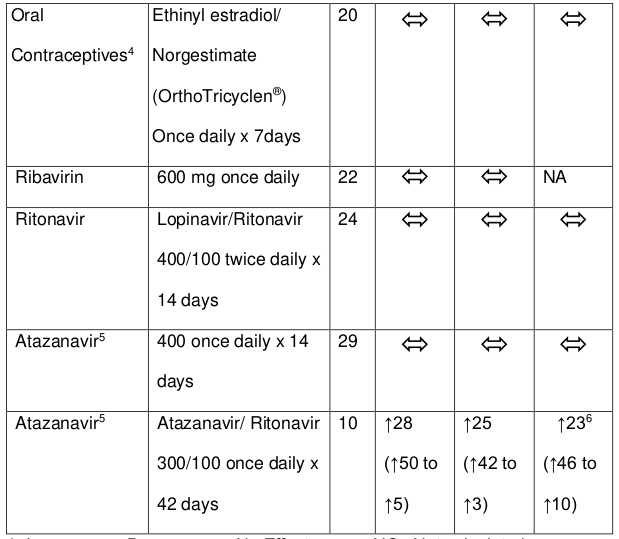
Table 2. Medicine interactions: Changes in pharmacokinetic parameters for co-administered medicine in the presence of tenofovir:
1 Increase =↑; Decrease =↓; No Effect = ; NC =Not calculated
2 R-(active), S-and total methadone exposures were equivalent when dosed alone or with tenofovir as tenofovir disoproxil fumarate 300 mg.
3 Individual patients were maintained on their stable methadone dose. No pharmacodynamic alterations (opiate toxicity or withdrawal signs or symptoms) were reported.
4 Ethinyl oestradiol and 17-deacetyl norgestimate (pharmacologically active metabolite) exposures where equivalent when dosed alone or with tenofovir as tenofovir disoproxil fumarate 300 mg.
5 RSA Innovator Volutrip Prescribing Information
6 In HIV infected patients, addition of tenofovir disoproxil fumarate to atazanavir 300 mg plus ritonavir 100 mg, resulted in AUC and Cmin values of atazanavir that were 2,3 and 4-fold higher than the respective values observed for atazanavir 400 mg when given alone.
The likelihood of metabolic interactions is low due to limited metabolism and plasma protein binding and almost complete renal clearance.
Zidovudine plasma levels are not significantly altered when co-administered with DOLTELAM. Zidovudine has no effect on the pharmacokinetics of DOLTELAM.
Co-administration of zidovudine results in a 13% increase in zidovudine exposure and 28% increase in peak plasma levels. This is not considered to be of significance to patient safety and therefore no dosage adjustments are necessary.
Table 3. Medicine interactions study reports with lamivudine:
DOLTELAM may inhibit the intracellular phosphorylation of zalcitabine when the two medicines are used concurrently. DOLTELAM is therefore not recommended to be used in combination with zalcitabine.
Administration of trimethoprim, a constituent of co-trimoxazole causes an increase in DOLTELAM plasma levels. Unless the patient has renal impairment, no dosage adjustment of DOLTELAM is necessary. DOLTELAM has no effect on the pharmacokinetics of co-trimoxazole.
The possibility of interactions with other medicines administered concurrently should be considered, particularly when the main route is renal.
The co-administration of DOLTELAM with etravirine (ETR) is not recommended unless the patient is also receiving concomitant atazanavir + ritonavir (ATV + RTV), lopinavir + ritonavir (LPV + RTV) or darunavir + ritonavir (DRV + RTV).
Rifampicin decreases the blood levels of dolutegravir. A supplementary dose of dolutegravir should be given to patients taking DOLTELAM. There is evidence that the concentration of isoniazid is increased by dolutegravir, as contained in DOLTELAM.
In vitro, dolutegravir as in DOLTELAM, demonstrated no direct, or weak inhibition (lC50 >50 µM) of the enzymes cytochrome P450 (CYP)1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A, uridine diphosphate glucuronosyl transferase (UGT)1A1 or UGT2B7, or the transporters Pgp, BCRP, OATP1B1, OATP1B3, OCT1 or MRP2.
In vitro, dolutegravir as in DOLTELAM did not induce CYP1A2, CYP2B6 or CYP3A4. In vivo, dolutegravir did not have an effect on midazolam, a CYP3A4 probe. Based on these data, dolutegravir is not expected to affect the pharmacokinetics of medicines that are substrates of these enzymes or transporters (e.g., reverse transcriptase and protease inhibitors, opioid analgesics, antidepressants, statins, azole antifungals (such as fluconazole, itraconazole, clotrimazole), proton pump inhibitors (such as esomeprazole, lansoprazole, omeprazole), antierectile dysfunction medicines (such as sildenafil, tadalafil, vardenafil), aciclovir, valaciclovir, sitagliptin, adefovir). In medicines interaction study reports, dolutegravir did not have a clinically relevant effect on the pharmacokinetics of the following: tenofovir, methadone, efavirenz, lopinavir, atazanavir, darunavir, etravirine, fosamprenavir, rilpivirine, telaprevir and oral contraceptives containing norgestimate and ethinyl estradiol.
In vitro, dolutegravir as in DOLTELAM inhibited the renal organic cation transporter 2 (OCT2). Based on this report, dolutegravir may increase plasma concentrations of medicines in which excretion is dependent upon OCT2 (dofetilide, metformin) (see Table 4).
Effect of other medicines on the pharmacokinetics of dolutegravir Dolutegravir, as in DOLTELAM, is eliminated mainly through metabolism by UGT1A1. It is also a substrate of UGT1A3, UGT1A9, CYP3A4, Pgp, and BCRP; therefore, medicines that induce those enzymes, may theoretically decrease dolutegravir plasma concentration and reduce the therapeutic effect of dolutegravir. Co-administration of DOLTELAM and other medicines that inhibit UGT1A1, UGT1A3, UGT1A9, CYP3A4, and/or Pgp may increase dolutegravir plasma concentration (see Table 4).
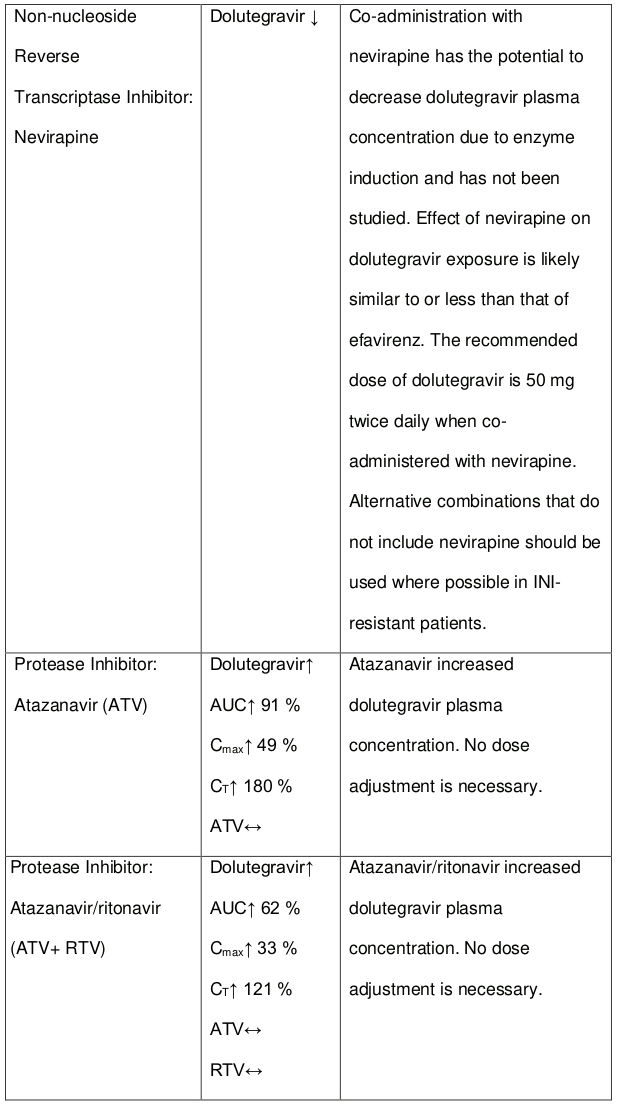
Efavirenz, nevirapine, rifampicin and tipranavir in combination with ritonavir each reduced the plasma concentrations of dolutegravir, as in DOLTELAM, significantly and require dolutegravir dose adjustment to 50 mg twice daily. Etravirine also reduced plasma concentrations, but the effect of etravirine was mitigated by co-administration of the CYP3A4 inhibitors lopinavir/ritonavir, darunavir/ritonavir and is expected to be mitigated by atazanavir/ritonavir. Therefore, no DOLTELAM dose adjustment is necessary when co-administered with etravirine and either Iopinavir/ritonavir, darunavir/ritonavir, or atazanavir/ritonavir. Another inducer, fosamprenavir in combination with ritonavir decreased plasma concentrations of dolutegravir but does not require a dosage adjustment of DOLTELAM. Caution is warranted and clinical monitoring is recommended when these combinations are given in INI-resistant patients (see Table 4: Medicine Interactions – HIV-1 Antiviral Medicines). A medicine interaction study with the UGT1A1 inhibitor, atazanavir, did not result in a clinically meaningful increase in the plasma concentrations of dolutegravir. Tenofovir, ritonavir, lopinavir/ritonavir, darunavir/ritonavir, rilpivirine, bocepravir, telaprevir, prednisone, rifabutin, and omeprazole had no or a minimal effect on dolutegravir pharmacokinetics, therefore no DOLTELAM dose adjustment is required when co-administered with these medicines.
Table 4. Medicine interactions:
Abbreviations: ↑ = increase; ↓ = decrease; ↔ = no significant change; AUC = area under the concentration versus time curve; Cmax = maximum observed concentration; CT = concentration at the end of dosing interval.
DOLTELAM should not be co-administered with polyvalent cation-containing antacids. It is recommended to be administered 2 hours before or 6 hours after these medicines (see section 4.5).
DOLTELAM may increase metformin concentrations therefore, metformin is contraindicated in patients taking DOLTELAM (see section 4.3).
DOLTELAM should not be prescribed in women who plan to become pregnant. Women of childbearing age should not use DOLTELAM unless they are using highly effective contraception.
Treatment with DOLTELAM should not be initiated without a medically supervised negative pregnancy test. This test should be repeated at frequent intervals during treatment with DOLTELAM and especially in the event that pregnancy is suspected.
DOLTELAM is contraindicated in pregnancy. Neural tube defects have been noted in an observational study in humans, where DTG-bases regimens were used at the time of conception and early pregnancy (see section 4.3).
Tenofovir, dolutegravir and lamivudine were shown to cross the placenta in reproductive toxicity studies in animals. Late onset neurological disorders, including seizures, have been observed in children who have been exposed to nucleoside analogues in utero such as tenofovir and lamivudine, (see Mitochondrial Dysfunction under section 4.4)
DOLTELAM is contraindicated in lactation. Mothers breastfeeding their infants should not use DOLTELAM. Lamivudine is excreted in human milk at similar concentrations to those found in serum; tenofovir is excreted in breast milk and it is not known whether dolutegravir is excreted in human milk.
There are no data on dolutegravir’s effects on human male or female fertility, although animal studies indicate no harmful effects of dolutegravir, lamivudine and tenofovir disoproxil on fertility.
DOLTELAM may affect the ability to drive and use machines as DOLTELAM can cause dizziness. Patients should ensure that they do not engage in driving or using machines until they know how DOLTELAM affects them.
The most severe adverse reactions linked to dolutegravir treatment are hypersensitivity reactions that include rash and severe liver effects. The most common adverse reactions of dolutegravir are nausea, diarrhoea and headache.
Renal impairment, renal failure and proximal renal tubulopathy (including Fanconi syndrome) sometimes leading to bone abnormalities (infrequently contributing to fractures) have been reported in patients receiving tenofovir disoproxil. Monitoring of renal function is recommended for patients receiving DOLTELAM (see section 4.4). The table below shows all adverse drug reactions (ADRs) observed during clinical trials and postmarket spontaneous reports with DOLTELAM
Frequency estimate: Frequent (≥1/100), Less frequent (<1/100), Not known (cannot be estimated from the available data).
Table 5. Adverse effects for DOLTELAM:
| System Organ Class | Frequency | ||
|---|---|---|---|
| Frequent | Less Frequent | Not known | |
| Blood and lymphatic system disorders | Neutropenia, anaemia, thrombocytopenia, pure red cell aplasia | ||
| Immune system disorders | Hypersensitivity, immune reactivation syndrome | ||
| Metabolism and nutrition disorders | Hypophosphatemia | Lactic acidosis | Hypokalaemia |
| Psychiatric disorders | Insomnia, abnormal dreams, depression, anxiety | Suicidal ideation or suicide attempt | |
| Nervous system disorders | Headache, dizziness | Peripheral neuropathy paraesthesia | |
| Respiratory, thoracic and mediastinal disorders | Cough, nasal symptoms | Dyspnoea | |
| Gastrointestinal disorders | Nausea, diarrhoea, vomiting, flatulence, upper abdominal pain, abdominal pain, abdominal discomfort | Pancreatitis, elevated serum amylases | |
| Hepatobiliary disorders | Hepatitis | Hepatic steatosis | |
| Skin and subcutaneous tissue disorders | Rash, pruritus, hair loss | ||
| Musculoskeletal and connective tissue disorders | Arthralgia, myalgia | Rhabdomyolysis, osteomalacia (manifested as bone pain and infrequently contributing to fractures), muscular weakness, osteonecrosis | |
| Renal and urinary disorders | Rare acute renal failure, renal failure, proximal renal tubulopathy (including Fanconi syndrome), increased serum creatinine, acute tubular necrosis | Nephritis (including acute interstitial nephritis), nephrogenic diabetes insipidus | |
| General disorders and administration site conditions | Fatigue, malaise, fever | Asthenia | Immune reconstitution syndrome |
| Investigations | Raised alanine aminotransferase (ALT) and aspartate aminotransferase (AST) raised creatine kinase | ||
Table 6. Side effects for Dolutegravir:
| System Organ Class | Frequency | ||
|---|---|---|---|
| Frequent | Less Frequent | Not known | |
| Immune system disorders | Hypersensitivity, immune reconstitution syndrome | ||
| Psychiatric disorders | Insomnia | ||
| Nervous system disorders | Headache, dizziness, abnormal dreams | ||
| Gastrointestinal disorders | Nausea, diarrhoea | Vomiting, flatulence, upper abdominal pain | Abdominal pain, abdominal discomfort |
| Hepatobiliary disorders | Hepatitis | ||
| Skin and subcutaneous tissue disorders | Rash, pruritus | ||
Table 7. Side effects for Lamivudine:
| System Organ Class | Frequency | ||
|---|---|---|---|
| Frequent | Less Frequent | Not known | |
| Blood and lymphatic system disorders | Neutropenia, anaemia, thrombocytopenia | Pure red cell aplasia | |
| Metabolism and nutrition disorders | Hyperlactataemia | Lactic acidosis, lipodystrophy | |
| Nervous system disorders | Headache, insomnia | Peripheral neuropathy (or paraesthesia), late onset neurological disorders in children exposed in utero | |
| Gastrointestinal disorders | Nausea, diarrhoea, vomiting, upper abdominal pain or cramps, stomatitis | Pancreatitis, rises in serum amylase | |
| Hepatobiliary disorders | Transient rises in liver enzymes (AST, ALT) | ||
| Skin and subcutaneous tissue disorders | Rash, alopecia | ||
| Musculoskeletal and connective tissue disorders | Arthralgia, muscle disorders | Rhabdomyolysis, decrease in bone mineral density, osteopenia, fractures | |
| General disorders and administration site conditions | Fatigue, malaise, fever | ||
Table 8. Side effects for Tenofovir disoproxil fumarate:
| System Organ Class | Frequency | ||
|---|---|---|---|
| Frequent | Less Frequent | Not known | |
| Immune system disorders | Allergic reaction | ||
| Metabolism and nutrition disorders | Hypophosphataemia, lactic acidosis | ||
| Respiratory, thoracic and mediastinal disorders | Dyspnoea | ||
| Gastrointestinal disorders | Abdominal pain, anorexia, dyspepsia, flatulence | Increased amylase, pancreatitis | |
| Hepatobiliary disorders | Increased liver enzymes, hepatitis | ||
| Renal and urinary disorders | Renal insufficiency, renal failure, proximal tubulopathy, proteinuria, increased creatinine, acute tubular necrosis, nephrogenic, diabetes insipidus | ||
Serum creatinine can increase in the first week of treatment with dolutegravir and then remain stable. A mean change from baseline of 10 μmol/litre occurred after 48 weeks of treatment. Creatinine increases were comparable between various background regimens. These changes are not considered clinically relevant since they do not reflect a change in glomerular filtration rate.
In HIV patients with severe immune deficiency at the start of combination antiretroviral therapy (CART), an inflammatory reaction to asymptomatic or residual opportunistic infections may arise. Autoimmune disorders (such as Graves' disease) have also been reported; however, the time to onset is more variable and these events can occur many months after starting treatment (see section 4.4).
As lamivudine and tenofovir disoproxil may cause renal damage, monitoring of renal function is recommended (see section 4.4). Proximal renal tubulopathy generally resolved or improved after tenofovir disoproxil discontinuation. However, in some patients, declines in creatinine clearance did not completely resolve despite tenofovir disoproxil discontinuation. Patients at risk of renal impairment (such as patients with baseline renal risk factors, advanced HIV disease, or patients receiving concomitant nephrotoxic medicines) are at increased risk of experiencing incomplete recovery of renal function despite tenofovir disoproxil discontinuation (see section 4.4).
The following adverse reactions, listed under the body system headings above, may occur as a consequence of proximal renal tubulopathy: rhabdomyolysis, osteomalacia (manifested as bone pain and infrequently contributing to fractures), hypokalaemia, muscular weakness, myopathy and hypophosphatemia. These events are not likely to be causally associated with tenofovir disoproxil therapy in the absence of proximal renal tubulopathy.
Co-administration of tenofovir disoproxil and didanosine is not recommended as it results in a 40-60% increase in systemic exposure to didanosine that may increase the risk of didanosine-related adverse reactions. (see section 4.5). Pancreatitis and lactic acidosis, sometimes fatal, have been reported.
In clinical studies with dolutegravir, the side effects profile in patients also infected with hepatitis B or C or both was similar to that in patients without hepatitis, provided that the baseline liver function tests did not exceed 5 times the upper limit of normal. However, the rates of AST and ALT abnormalities were higher in patients with hepatitis B or C co-infection. Liver enzymes elevations consistent with immune reactivation syndrome occurred in some subjects with hepatitis B or C co-infection at the start of dolutegravir therapy, particularly in those whose hepatitis B therapy was stopped.
Limited data on patients co-infected with HIV/HBV or HIV/HCV indicate that the adverse reaction profile of emtricitabine and tenofovir disoproxil in patients co-infected with HIV/HBV or HIV/HCV was similar to that observed in patients infected with HIV without co-infection. However, as would be expected, elevations in AST and ALT occurred more frequently than in the general HIV infected population.
In HIV infected patients co-infected with HBV, clinical and laboratory evidence of hepatitis may occur after discontinuation of treatment (see section 4.4).
The limited data available for children and adolescents (aged 6 to 18 years and weighing at least 15 kg) using dolutegravir suggest no additional adverse reactions beyond those that occur in adults. The adverse reactions observed in paediatric patients who received treatment with tenofovir disoproxil or lamivudine as single entities were consistent with those observed in clinical studies in adults.
Reductions in bone mineral density (BMD) have been reported with tenofovir disoproxil in paediatric patients. In HIV-infected adolescents, the BMD Z-scores in subjects who received tenofovir disoproxil were lower than those in subjects who received placebo. In HIV-infected children, the BMD Z-scores in subjects who switched to tenofovir disoproxil were lower than those in subjects who remained on regimens containing stavudine or zidovudine.
Caution should be exercised since elderly patients are more likely to have decreased renal function.
Reporting suspected adverse reactions after authorisation of the medicine is important. It allows continued monitoring of the benefit/risk balance of the medicine. Healthcare professionals are asked to report any suspected adverse reactions to SAHPRA via the “6.04 Adverse Drug Reaction Reporting Form”, found online under SAHPRA’s publications: https://www.sahpra.org.za/Publications/Index/8.
Not applicable.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.