EVRENZO Film-coated tablet Ref.[27944] Active ingredients: Roxadustat
Source: European Medicines Agency (EU) Revision Year: 2021 Publisher: Astellas Pharma Europe B.V., Sylviusweg 62, 2333 BE Leiden, The Netherlands
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: anti-anaemic preparations, other anti-anaemic preparations
ATC code: B03XA05
Mechanism of action
Roxadustat is a hypoxia-inducible factor, prolyl hydroxylase inhibitor (HIF-PHI). The activity of HIF-PH enzymes controls intracellular levels of HIF, a transcription factor that regulates the expression of genes involved in erythropoiesis. Activation of the HIF pathway is important in the adaptative response to hypoxia to increase red blood cell production. Through the reversible inhibition of HIF-PH, roxadustat stimulates a coordinated erythropoietic response that includes the increase of plasma endogenous erythropoietin (EPO) levels, regulation of iron transporter proteins and reduction of hepcidin (an iron regulator protein that is increased during inflammation in CKD). This results in improved iron bioavailability, increased Hb production and increased red cell mass.
Pharmacodynamic effects
Effects on QTc and heart rate
A thorough QT (TQT) study in healthy subjects with roxadustat at a single therapeutic dose of 2.75 mg/kg and a single supratherapeutic dose of 5 mg/kg (up to 510 mg) did not show a prolongation of the QTc interval. The same thorough QT study demonstrated a placebo-corrected heart rate increase of up to 9 to 10 bpm at 8 to 12 h post-dose for the 2.75 mg/kg dose and 15 to 18 bpm at 6 to 12 h post-dose for the dose of 5 mg/kg.
Clinical efficacy and safety
Development program in anaemia with CKD
Efficacy and safety of roxadustat were evaluated for at least 52 weeks in a globally conducted phase 3 program comprising of 8 multicentre and randomized studies in non-dialysis dependent (NDD) and dialysis-dependent (DD) CKD patients with anaemia (see Table 4).
Three studies in stage 3-5 CKD NDD patients were double-blind and placebo-controlled studies (ALPS, 1517-CL-0608; ANDES, FGCL-4592-060; OLYMPUS, D5740C00001) and one study was open-label ESA-controlled (DOLOMITES, 1517-CL-0610) using darbepoetin alfa as comparator. All NDD studies assessed efficacy and safety in ESA-untreated patients by correcting and thereafter maintaining Hb in the target range of 10 to 12 g/dL (Hb correction setting).
Four open-label ESA-controlled DD studies (control: epoetin alfa and/or darbepoetin alfa) in patients on haemodialysis or peritoneal dialysis assessed the efficacy and safety in different settings:
- in a Hb correction setting (HIMALAYAS, FGCL-4592-063).
- in an ESA conversion setting converting patients from treatment with an ESA to maintain Hb in the target range (PYRENEES, 1517-CL-0613; SIERRAS, FGCL-4592-064).
- or combining the Hb correction and ESA conversion approaches (ROCKIES, D5740C00002).
Patients in the NDD studies had CKD stage 3 to 5 and were not receiving dialysis. All patients had an average Hb ≤10.0 g/dL except patients in the DOLOMITES study (1517-CL-0610), which allowed an average Hb ≤10.5 g/dL. Ferritin levels were required to be ≥30 ng/mL (ALPS, 1517-CL-0608; ANDES, FGCL-4592-060), ≥50 ng/mL (OLYMPUS, D5740C00001) or ≥100 ng/mL (DOLOMITES, 1517-CL-0610). Except for those in the (OLYMPUS, D5740C00001) study, which allowed ESA treatment until 6 weeks prior to randomization, patients could not have received any ESA treatment within 12 weeks of randomization.
Patients in the DD studies had to be on dialysis: stable DD for patients in the PYRENEES study (1517-CL-0613), which was defined as dialysis for longer than 4 months; or incident (ID), DD for patients in the HIMALAYAS study (FGCL-4592-063), which was defined as dialysis ≥2 weeks but ≤4 months. Patients in the SIERRAS (FGCL-4592-064) and ROCKIES studies (D5740C00002) included both stable (approximately 80% to 90%) and ID (approximately 10% to 20%) DD patients. Ferritin was required to be ≥100 ng/mL in all patients. All patients required intravenous or subcutaneous ESA for at least 8 weeks prior to randomization, except those patients in the HIMALAYAS study (FGCL-4592-063) which excluded patients who had received any ESA treatment within 12 weeks prior to randomization.
Treatment with roxadustat followed the principles of dosing instructions as described in section 4.2. Demographics and all baseline characteristics across individual studies were comparable between the roxadustat and control groups. The median age at randomization was 55 to 69 years, with between 16.6% and 31.1% in the 65-74 age range, and between 6.8% and 35% who were ≥75 years of age. The percentage of female patients ranged from 40.5% to 60.7%. The most commonly represented races across the studies were White, Black or African American and Asian. The most common CKD aetiologies were diabetic and hypertensive nephropathy. Median Hb levels ranged from 8.60 to 10.78 g/dL. Approximately 50-60% of NDD patients and 80-90% of DD patients were iron replete at baseline.
Data from seven phase 3 studies were pooled in two separate populations (three NDD and four DD) (see Table 4).
Three placebo-controlled NDD Studies (2,386 patients on roxadustat; 1,884 patients on placebo) were included in the NDD pool. Data from the phase 3 ESA-controlled NDD DOLOMITES study (1517-CL-0610; 323 patients on roxadustat and 293 patients on darbepoetin alfa) are not included in the NDD pooled analyses as this study is the only open-label, active-controlled study in the NDD population.
Four ESA-controlled DD Studies (2,354 patients on roxadustat; 2,360 patients on ESA [epoetin alfa and/or darbepoetin alfa]) were included in the DD pool. Within the DD pool, two sub pools were established to reflect the two different treatment settings:
- Patients in the DD population who were on dialysis for greater than 2 weeks and less than 4 months were termed incident (ID) DD patients (ID DD pool) reflective of the Hb correction setting.
- The DD patients who were on dialysis after this threshold of four months were termed stable DD patients (Stable DD pool) reflective of the ESA conversion setting.
Table 4. Overview on Roxadustat phase 3 development program in anaemia with CKD:
| Studies in NDD patients | ||||
|---|---|---|---|---|
| Placebo-controlled studies (NDD pool) | ESA-control (Darbepoetin alfa) | |||
| Setting | Hb correction | |||
| Study | ALPS (1517-CL-0608) | ANDES (FGCL-4592-060) | OLYMPUS (D5740C00001) | DOLOMITES (1517-CL-0610) |
| Randomized (roxadustat/comparator) | 594 (391/203) | 916 (611/305) | 2760 (1384/1376) | 616 (323/293) |
| Studies in DD patients | ||||
| ESA-controlled studies (DD pool) (Epoetin alfa or Darbepoetin alfa) | ||||
| Setting | ESA conversion | Hb correction | ESA conversion and Hb correction | |
| Study | PYRENEES (1517-CL-0613) | SIERRAS (FGCL-4592-064) | HIMALAYAS (FGCL-4592-063) | ROCKIES (D5740C00002) |
| Randomized (roxadustat/comparator) | 834 (414/420) | 740 (370/370) | 1039 (522/517) | 2101 (1048/1053) |
DD: dialysis dependent; ESA: erythropoiesis-stimulating agent; Hb: haemoglobin;
NDD: non-dialysis dependent.
NDD CKD patients
Efficacy results
Course of Hb during treatment
In clinical studies, roxadustat was effective in achieving and maintaining target Hb levels (10-12 g/dL) in patients with CKD anaemia not on dialysis (see Figure 1).
Figure 1. Mean (SE) Hb (g/dL) over time up to week 52 (FAS); NDD pool (Hb correction):
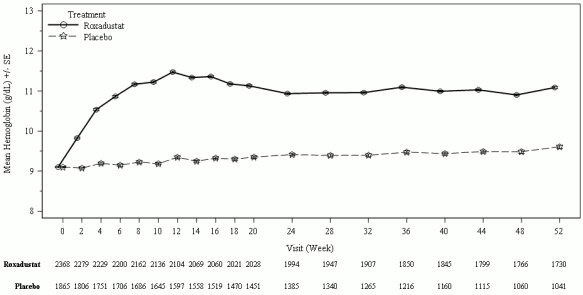
FAS: full analysis set; Hb: haemoglobin; NDD: non-dialysis dependent; SE: standard error.
Key Hb efficacy endpoints in NDD CKD patients
In NDD patients in need of anaemia treatment for Hb correction, the proportion of patients who achieved Hb response during the first 24 weeks was higher in the roxadustat group (80.2%) compared with placebo (8.7%). There was a statistically significant increase in Hb from baseline to weeks 28 to 36 in the roxadustat group (1.91 g/dL) compared with placebo (0.14 g/dL) and the lower limit of the 95% confidence interval is above 1. In the NDD studies, an increase in Hb of at least 1 g/dL was achieved with a median time of 4.1 weeks (see Table 5).
In the open-label ESA-controlled NDD DOLOMITES (1517-CL-0610) study, the proportion of patients who achieved Hb response during the first 24 weeks was non-inferior in the roxadustat group (89.5%) compared with darbepoetin alfa (78%) (see Table 5).
Table 5. Key Hb efficacy endpoints (NDD):
| Population | NDD CKD patients | |||
|---|---|---|---|---|
| Setting | Hb correction | Hb correction | ||
| Endpoint/Parameter | NDD pool (FAS) | DOLOMITES (PPS) 1517-CL-0610 | ||
| Roxadustat n=2368 | Placebo n=1865 | Roxadustat n=286 | Darbepoetin alfa n=273 | |
| Proportion of patients who achieved Hb response1 | ||||
| Responders, n ( % ) [95% CI] | 1,899 (80.2) [78.5, 81.8] | 163 (8.7) [7.5, 10.1] | 256 (89.5) [85.4, 92.8] | 213 (78.0) [72.6, 82.8] |
| Difference of proportions [95% CI] | 71.5 [69.40, 73.51] | 11.51 [5.66, 17.36] | ||
| Odds ratio [95% CI] | 40.49 [33.01, 49.67] | 2.48 [1.53, 4.04] | ||
| P value | <0.0001 | ND | ||
| Change from baseline in Hb (g/dL)2 | ||||
| Mean (SD) baseline | 9.10 (0.74) | 9.10 (0.73) | 9.55 (0.76) | 9.54 (0.69) |
| Mean (SD) CFB | 1.85 (1.07) | 0.17 (1.08) | 1.85 (1.08) | 1.84 (0.97) |
| LS mean | 1.91 | 0.14 | 1.85 | 1.84 |
| LS mean difference [95% CI] | 1.77 [1.69, 1.84] | 0.02 [-0.13, 0.16] | ||
| P value | <0.0001 | 0.844 | ||
CFB: change from baseline; CI: confidence interval; CKD: chronic kidney disease; FAS: full analysis set; Hb: haemoglobin; LS: Least squares; ND: not done; NDD: non-dialysis dependent; PPS: per protocol set; SD: standard deviation.
1 Hb response within the first 24 weeks
2 Change from baseline in Hb to Weeks 28 to 36
DD CKD patients
Course of Hb during treatment
In clinical studies, roxadustat was effective in achieving and maintaining target Hb levels (10-12 g/dL) in CKD patients on dialysis, irrespective of prior ESA treatment (see Figures 2 and 3).
Figure 2. Mean (SE) Hb up to week 52 (FAS); ID DD subpool (Hb correction):
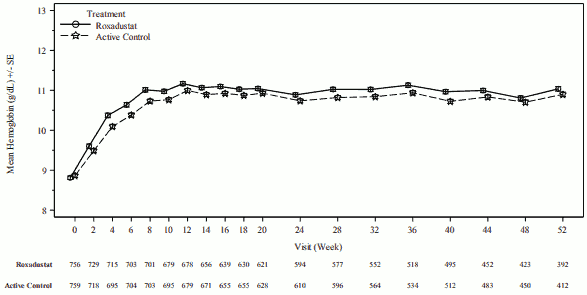
DD: dialysis-dependent; FAS: full analysis set; Hb: haemoglobin; ID: incident; SE: standard error.
Figure 3. Mean (SE) Hb (g/dL) over time up to week 52 (FAS); stable DD subpool (ESA conversion):

DD: dialysis dependent; ESA: erythropoiesis-stimulating agent; FAS: full analysis set; Hb: haemoglobin; SE: standard error.
Key Hb efficacy endpoints in DD CKD patients
In DD patients in need of anaemia treatment for Hb correction and those converted from ESA treatment, there was an increase in Hb from baseline to weeks 28 to 36 in the roxadustat group; this increase was comparable to that observed in the ESA group and was above the prespecified noninferiority margin of -0.75 g/dL. The proportion of patients who achieved Hb response during the first 24 weeks was similar in the roxadustat and ESA groups (see Table 6).
Table 6. Key Hb efficacy endpoints (DD):
| Population | DD Patients | |||
|---|---|---|---|---|
| Setting | Hb Correction | ESA Conversion | ||
| Endpoint/Parameter | ID DD pool (FAS/PPS) | Stable DD Pool (PPS) | ||
| Roxadustat n=756 | ESA n=759 | Roxadustat n=1379 | ESA n=1417 | |
| Change from baseline in Hb (g/dL) | ||||
| Mean (SD) baseline | 8.77 (1.20) | 8.82 (1.20) | 10.32 (0.99) | 10.37 (0.99) |
| Mean (SD) CFB | 2.37 (1.57) | 2.12 (1.46) | 0.65 (1.15) | 0.36 (1.23) |
| LS mean | 2.17 | 1.89 | 0.58 | 0.28 |
| LS mean difference [95% CI] | 0.28 [0.110, 0.451] | 0.30 [0.228, 0.373] | ||
| P value | 0.0013 | <0.0001 | ||
| Proportion of patients who achieved Hb response1,2 | ||||
| Responders, n ( % ) [95% CI] | 453 (59.9) [56.3, 63.4] | 452 (59.6) [56.0, 63.1] | 978 (70.9) [68.4, 73.3] | 959 (67.7) [65.2, 70.1] |
| Difference of proportions [95% CI] | 0.3 [-4.5, 5.1] | 2.7 [-0.7, 6.0] | ||
| Odds ratio [95% CI] | ND | ND | ||
| P value | ND | ND | ||
CFB: change from baseline; CI: confidence interval; CKD: chronic kidney disease; DD: dialysisdependent; ESA: erythropoiesis-stimulating agent; FAS: full analysis set; Hb: haemoglobin; ID: incident; LS: Least squares; ND: not done; PPS: per protocolset; SD: standard deviation.
1 Hb within the target range of 10.0 to 12.0 g/dL during weeks 28 to 36 without having received rescue therapy within 6 weeks prior to and during this 8-week evaluation period.
2 Data in the ID DD pool were only analysed for weeks 28 to 52.
Rescue therapy, RBC transfusion and intravenous iron
The effects of treatment with roxadustat on use of rescue therapy, RBC transfusion and intravenous iron are presented in Table 7 (NDD) and Table 8 (DD). In clinical studies, roxadustat reduced hepcidin (regulator of iron metabolism), reduced ferritin, increased serum iron while transferrin saturation was stable, all which were assessed over time as indicators of iron status.
Low-density lipoprotein (LDL) cholesterol
The effects of treatment with roxadustat on LDL cholesterol are presented in Tables 7 and 8. There was a reduction in mean LDL and high density lipoprotein (HDL) cholesterol levels in roxadustattreated patients compared with placebo or ESA-treated patients. The effect on LDL cholesterol was more pronounced, leading to a reduction of the LDL/HDL ratio and was observed regardless of the use of statins.
Table 7. Other efficacy endpoints: use of rescue therapy, monthly intravenous iron use and change from baseline in LDL cholesterol (NDD):
| Population | NDD CKD patients | |||
|---|---|---|---|---|
| Intervention | Correction | Correction | ||
| Endpoint/Parameter | NDD pool (FAS) | DOLOMITES (1517-CL-0610) | ||
| Roxadustat n=2368 | Placebo n=1865 | Roxadustat n=322 | Darbepoetin alfa n=292 | |
| Number of patients with rescue therapy, n (%)1 | 211 (8.9) | 580 (31.1) | ND | |
| RBC | 118 (5.0) | 240 (12.9) | ||
| IV iron | 50 (2.1) | 90 (4.8) | ||
| ESA | 48 (2.0) | 257 (13.8) | ||
| IR | 10.4 | 41.0 | ||
| Hazard ratio | 0.19 | ND | ||
| 95% CI | 0.16, 0.23 | |||
| P value | <0.0001 | |||
| Number of Patients with IV Iron, n (%)2 | ND | 20 (6.2) | 37 (12.7) | |
| IR | 9.9 | 21.2 | ||
| Hazard ratio | 0.45 | |||
| 95% CI | 0.26, 0.78 | |||
| P value | 0.004 | |||
| Change from baseline in LDL cholesterol (mmol/L) to weeks 12 to 283 | ||||
| Analysis using ANCOVA | ||||
| LS mean | -0.446 | 0.066 | -0.356 | 0.047 |
| 95% CI | -0.484, -0.409 | 0.017, 0.116 | -0.432, -0.280 | -0.033, 0.127 |
| LS mean difference (R-comparator) | -0.513 | -0.403 | ||
| 95% CI | -0.573, -0.453 | -0.510, -0.296 | ||
| P value | <0.0001 | <0.001 | ||
P values presented for the NDD pool are nominal p values.
ANCOVA: analysis of covariance; CI: confidence interval; ESA: erythropoiesis-stimulating agent; FAS: full analysis set; IR: incidence rate (per 100 patient-years at risk); IV: intravenous;
LDL: low density lipoprotein; LS: least squares; ND: not done; NDD: non-dialysis-dependent;
R: roxadustat; RBC: red blood cell;
1 For use of rescue therapy the NDD pool was analysed up to week 52.
2 During weeks 1-36.
3 Change from baseline in LDL cholesterol was assessed only through week 24 for study OLYMPUS (D5740C00001).
Table 8. Other efficacy endpoints: use of rescue therapy, monthly intravenous iron use and change from baseline in LDL cholesterol (DD):
| Population | DD CKD patients | |||
|---|---|---|---|---|
| Intervention | Correction | Conversion | ||
| Endpoint/Parameter | ID DD pool (FAS) | Stable DD pool (FAS) | ||
| Roxadustat n=756 | ESA n=759 | Roxadustat n=1586 | ESA n=1589 | |
| Mean monthly IV iron over weeks 28-52 (mg)1 | ||||
| n | 606 | 621 | 1414 | 1486 |
| Mean (SD) | 53.57 (143.097) | 70.22 (173.33) | 42.45 (229.80) | 61.99 (148.02) |
| Change from baseline in LDL cholesterol (mmol/L) to weeks 12 to 28 | ||||
| Analysis using ANCOVA | ||||
| LS mean | -0.610 | -0.157 | -0.408 | -0.035 |
| 95% CI | -0.700, -0.520 | -0.245, -0.069 | -0.449, -0.368 | -0.074, 0.003 |
| LS mean difference (R-comparator) | -0.453 | -0.373 | ||
| 95% CI | -0.575, -0.331 | -0.418, -0.328 | ||
| P value | <0.0001 | <0.0001 | ||
P values presented for the ID DD and stable DD pools are nominal p values. ANCOVA: analysis of covariance; CI: confidence interval; CKD: chronic kidney disease; DD: dialysis-dependent; ESA: erythropoiesis-stimulating agent; FAS: full analysis set; ID: incident dialysis; IV: intravenous; LDL: low density lipoprotein; LS: least squares; R: roxadustat.
1 Time period for PYRENEES (1517-CL-0613) study was up to week 36, and the time period for ROCKIES (D5740C0002) study was from week 36 through end of study.
In the dialysis study SIERRAS (FGCL-4592-064) a significantly lower proportion of patients received a red blood cell transfusion during treatment in the roxadustat group compared with the EPO-alfa group (12.5% versus 21.1%); the numerical reduction was not statistically significant in the ROCKIES (D5740C00002) study (9.8% versus 13.2%).
Patient reported outcomes not on dialysis
In the DOLOMITES study (1517-CL-0610) noninferiority of roxadustat to darbepoetin was established with regards to SF-36 PF and SF-36 VT.
Patient reported outcomes on dialysis
In the PYRENEES study (1517-CL-0613), non-inferiority of roxadustat to ESAs was established regarding SF-36 PF and SF-36 VT changes from baseline to weeks 12 to 28.
Clinical safety
Meta-analysis of pooled, adjudicated cardiovascular events
A meta-analysis, of adjudicated major adverse cardiovascular events (MACE; a composite of all-cause mortality [ACM], myocardial infarction, stroke) and MACE+ (a composite of ACM, myocardial infarction, stroke, and hospitalisation for either unstable angina or congestive heart failure), from the phase 3 study program was conducted in 8984 patients.
MACE, MACE+ and ACM outcomes are presented for three datasets using the pooled hazard ratio (HR) and its 95% confidence interval (CI). The three datasets include:
- A pooled placebo-controlled Hb correction dataset in NDD patients [includes patients from studies OLYMPUS (D5740C00001), ANDES (FGCL-4592-060) and ALPS (1517-CL-0608); see Table 4]
- A pooled ESA-controlled Hb correction dataset in NDD and ID-DD patients [includes patients from studies DOLOMITES (1517-CL-0610), HIMALAYAS (FGCL-4592-063), and the ID-DD patients of studies SIERRAS (FGCL-4592-064) and ROCKIES (D5740C00002); see Table 4]
- A pooled ESA-controlled ESA conversion dataset in Stable DD patients [includes patients from study PYRENEES (1517-CL-0613) and Stable DD patients from studies ROCKIES (D5740C00002) and SIERRAS (FGCL-4592-064); see Table 4]
MACE, MACE+ and ACM in the placebo-controlled Hb correction set of non-dialysis-dependent CKD patients
In NDD patients the analysis for MACE, MACE+ and ACM of the on-treatment analyses included all data from the start of study treatment until 28 days of the end of treatment follow-up. The on-treatment analyses used a Cox model weighted inversely for the probability of censoring (IPCW method) which aims to correct for follow-up time differences between roxadustat and placebo including identified contributors to increased risk and early discontinuation, in particular estimated glomerular filtration rate (eGFR) determinants and Hb at baseline and over time. Whether any residual confounding is present with this model remains uncertain. The HRs for the on-treatment analyses were 1.26, 1.17 and 1.16 (see Table 9). The ITT analyses included all data from the start of study treatment until the end of posttreatment safety follow-up. The ITT analysis has been included to illustrate an imbalance in risk distribution favouring placebo in the on-treatment analysis, however, ITT analyses generally demonstrate a dilution of study drug treatment effect and in these ITT analyses bias cannot be completely excluded, especially as ESA rescue therapy was introduced after study treatment discontinuation. The HRs were 1.10, 1.07 and 1.08, with upper limits of the 95% CIs of 1.27, 1.21 and 1.26, respectively.
Table 9. CV safety and mortality in placebo-controlled Hb correction NDD pool:
| MACE | MACE+ | ACM | ||||
|---|---|---|---|---|---|---|
| Roxadustat n=2386 | Placebo n=1884 | Roxadustat n=2386 | Placebo n=1884 | Roxadustat n=2386 | Placebo n=1884 | |
| On-treatment | ||||||
| Number of patients with events (%) | 344 (14.4) | 166 (8.8) | 448 (18.8) | 242 (12.8) | 260 (10.9) | 122 (6.5) |
| FAIR | 8.7 | 6.8 | 11.6 | 10.1 | 6.4 | 5.0 |
| HR (95% CI) | 1.26 (1.02, 1.55) | 1.17 (0.99, 1.40) | 1.16 (0.90, 1.50) | |||
| ITT | ||||||
| Number of patients with events (%) | 480 (20.1) | 350 (18.6) | 578 (24.2) | 432 (22.9) | 400 (16.8) | 301 (16) |
| FAIR | 10.6 | 10.3 | 13.2 | 13.2 | 8.3 | 8.1 |
| HR (95% CI) | 1.10 (0.96, 1.27) | 1.07 (0.94, 1.21) | 1.08 (0.93, 1.26) | |||
ACM: all-cause mortality; ACM is a component of MACE/MACE+. CI: confidence interval; FAIR: follow-up adjusted incidence rate (number of patients with event/100 patient years); HR: hazard ratio; ITT: intent-to-treat; MACE: major adverse cardiovascular event (death, non-fatal myocardial infarction and/or stroke); MACE+: major adverse cardiovascular event including hospitalisations for either unstable angina and/or congestive heart failure.
MACE, MACE+ and ACM in the ESA-controlled Hb correction set of non-dialysis-dependent and incident dialysis-dependent CKD patients
In the Hb correction setting of NDD and ID-DD patients baseline characteristics and treatment discontinuation rates were comparable between the pooled roxadustat and pooled ESA patients. The analysis for MACE, MACE+ and ACM observed on treatment showed HRs of 0.79, 0.78 and 0.78, with upper limits of the 95% CIs of 1.02, 0.98 and 1.05, respectively (see Table 10). The on-treatment analyses support no evidence of increased cardiovascular safety or mortality risk with roxadustat compared with ESA in CKD patients requiring Hb correction.
Table 10. CV safety and mortality in ESA-controlled Hb correction pool:
| MACE | MACE+ | ACM | ||||
|---|---|---|---|---|---|---|
| Roxadustat n=1083 | ESA n=1059 | Roxadustat n=1083 | ESA n=1059 | Roxadustat n=1083 | ESA n=1059 | |
| On-treatment | ||||||
| Number of patients with events (%) | 105 (9.7) | 136 (12.8) | 134 (12.4) | 171 (16.1) | 74 (6.8) | 99 (9.3) |
| IR | 6.5 | 8.2 | 8.3 | 10.3 | 4.6 | 6.0 |
| HR (95% CI) | 0.79 (0.61, 1.02) | 0.78 (0.62, 0.98) | 0.78 (0.57, 1.05) | |||
ACM: all-cause mortality; ACM is a component of MACE/MACE+, CI: confidence interval; ESA: erythropoiesis-stimulating agent; HR: hazard ratio; IR: incidence rate (number of patients with event/100 patient years); MACE: major adverse cardiovascular event (death, non-fatal myocardial infarction and/or stroke); MACE+: major adverse cardiovascular event including hospitalisations for either unstable angina and/or congestive heart failure.
MACE, MACE+ and ACM in ESA-controlled ESA conversion set of stable dialysis-dependent CKD patients
In stable DD patients converting from ESA analysis results for MACE, MACE+ and ACM observed on treatment showed HRs of 1.18, 1.03 and 1.23, with upper limits of the 95% CIs for HRs of 1.38, 1.19 and 1.49, respectively (see Table 11). The results in Table 11 should be interpreted with caution as patients allocated to roxadustat were switched from ESA at the start of the study and the impact of an inherent risk in switching to any new treatment versus remaining on a treatment with a stabilised Hb may confound the observed results and thus any comparison of treatment effect estimates cannot be reliably established.
Table 11. CV safety and mortality in ESA-controlled ESA conversion stable DD pool:
| MACE | MACE+ | ACM | ||||
|---|---|---|---|---|---|---|
| Roxadustat n=1594 | ESA n=1594 | Roxadustat n=1594 | ESA n=1594 | Roxadustat n=1594 | ESA n=1594 | |
| On-treatment | ||||||
| Number of patients with events (%) | 297 (18.6) | 301 (18.9) | 357 (22.4) | 403 (25.3) | 212 (13.3) | 207 (13.0) |
| IR | 10.4 | 9.2 | 12.5 | 12.3 | 7.4 | 6.3 |
| HR (95% CI) | 1.18 (1.00, 1.38) | 1.03 (0.90, 1.19) | 1.23 (1.02, 1.49) | |||
ACM: all-cause mortality; ACM is a component of MACE/MACE+. CI: confidence interval; ESA: erythropoiesis-stimulating agent; HR: hazard ratio; IR: incidence rate (number of patients with event/100 patient years); MACE: major adverse cardiovascular event (death, non-fatal myocardial infarction and/or stroke); MACE+: major adverse cardiovascular event including hospitalisations for either unstable angina and/or congestive heart failure.
5.2. Pharmacokinetic properties
Roxadustat plasma exposure (area under the plasma drug concentration over time curve [AUC] and maximum plasma concentrations [Cmax]) is dose-proportional within the recommended therapeutic dose range. In a three times per week dosing regimen, steady-state roxadustat plasma concentrations are achieved within one week (3 doses) with minimal accumulation. The pharmacokinetics of roxadustat do not change over time.
Absorption
Maximum plasma concentrations (Cmax) are usually achieved at 2 hours post dose in the fasted state. Administration of roxadustat with food decreased Cmax by 25% but did not alter AUC as compared with the fasted state. Therefore, roxadustat can be taken with or without food (see section 4.2).
Distribution
Roxadustat is highly bound to human plasma proteins (approximately 99%), predominantly to albumin. The blood-to-plasma ratio of roxadustat is 0.6. The apparent volume of distribution at steady state is 24 L.
Biotransformation
Based on in vitro data, roxadustat is a substrate for CYP2C8 and UGT1A9 enzymes, as well as BCRP, OATP1B1, OAT1 and OAT3. Roxadustat is not a substrate for OATP1B3 or P-gp. Roxadustat is primarily metabolised to hydroxy-roxadustat and roxadustat-O-glucuronide. Unchanged roxadustat was the major circulating component in human plasma; no detectable metabolite in human plasma constituted more than 10% of total drug-related material exposure and no human specific metabolites were observed.
Elimination
The mean effective half-life (t1/2) of roxadustat is approximately 15 hours in patients with CKD. The apparent total body clearance (CL/F) of roxadustat is 1.1 L/h in patients with CKD not on dialysis and 1.4 L/h in patients with CKD on dialysis. Roxadustat and its metabolites are not significantly removed by haemodialysis. When radiolabelled roxadustat was administered orally in healthy subjects, the mean recovery of radioactivity was 96% (50% in faeces, 46% in urine). In faeces, 28% of the dose was excreted as unchanged roxadustat. Less than 2% of the dose was recovered in urine as unchanged roxadustat.
Special Populations
Effects of age, sex, body weight, and race
No clinically relevant differences in the pharmacokinetics of roxadustat were observed based on age (≥18), sex, race, body weight, renal function (eGFR) or dialysis status in adult patients with anaemia due to CKD.
Haemodialysis
In dialysis-dependent CKD patients, no marked differences in pharmacokinetic parameter values were observed when roxadustat was administered 2 hours before or 1 hour after haemodialysis. Dialysis is a negligible route of overall clearance of roxadustat.
Hepatic impairment
Following a single dose of 100 mg roxadustat, mean roxadustat AUC was 23% higher and mean Cmax was 16% lower in subjects with moderate hepatic impairment (Child-Pugh Class B) and normal renal function compared to subjects with normal hepatic and renal functions. Subjects with moderate hepatic impairment (Child-Pugh Class B) and normal renal function showed an increase in unbound roxadustat AUCinf (+70%) as compared to healthy subjects. The pharmacokinetics of roxadustat in subjects with severe hepatic impairment (Child-Pugh Class C) have not been studied.
Drug-Drug Interactions
Based on in vitro data, roxadustat is an inhibitor of CYP2C8, BCRP, OATP1B1 and OAT3 (see section 4.5). The pharmacokinetics of rosiglitazone (moderate sensitive CYP2C8 substrate) were not affected by co-administration of roxadustat. Roxadustat may be an inhibitor of intestinal but not hepatic UGT1A1 and showed no inhibition of other CYP metabolising enzymes or transporters, or induction of CYP enzymes at clinically relevant concentrations. There is no clinically significant effect of oral adsorptive charcoal or omeprazole on roxadustat pharmacokinetics. Clopidogrel has no effect on roxadustat exposure in patients with CKD.
5.3. Preclinical safety data
Repeat-dose toxicity studies
In the 26-week intermittent repeat dose study in Sprague-Dawley or Fisher rats, roxadustat at approximately 4 to 6-fold the total AUC at Maximum Recommended Human Dose (MRHD) resulted in histopathological findings including aortic and atrioventricular valves (A-V) valvulopathies. These findings were present in surviving animals at the time of termination as well as in animals terminated early in a moribund state. Furthermore, the findings were not fully reversible as they were also present in animals at the end of a 30-day recovery period.
Exaggerated pharmacology resulting in excessive erythropoiesis has been observed in repeated-dose toxicity studies in healthy animals.
Haematological changes such as decreases in circulating platelets as well as increases in activated partial thromboplastin time and prothrombin time were noted in rats from approximately 2-fold the total AUC at MRHD. Thrombi were noted in the bone marrow (systemic exposures of approximately 7-fold the total AUC at MRHD in rats), kidneys (systemic exposures of approximately 5 to 6-fold total AUC at MRHD in rats), lungs (systemic exposures approximately 8- and 2-fold total AUC at MRHD in rats and cynomolgus monkeys, respectively), and the heart (systemic exposures of approximately 4 to 6-fold the total AUC at MRHD in rats).
Brain safety
In the 26-week intermittent repeat dose study in Sprague-Dawley rats, one animal, at approximately 6- fold the total AUC at MRHD showed a histologic finding of brain necrosis and gliosis. In Fisher rats, treated for the same duration, brain/hippocampal necrosis was noted in a total of four animals at the approximately 3 to 5-fold the total AUC at MRHD.
Cynomolgus monkeys intermittently administered roxadustat for 22 or 52-weeks, did not show similar findings at systemic exposures up to approximately 2-fold the total AUC at MRHD.
Carcinogenicity and mutagenicity
Roxadustat was negative in the in vitro Ames mutagenicity test, in vitro chromosome aberration test in human peripheral blood lymphocytes and an in vivo micronucleus test in mice at 40-fold the MRHD based on a human equivalent dose.
In the mouse and rat carcinogenicity studies, animals were administered roxadustat with the clinical dosing regimen of three times per week. Due to the rapid clearance of roxadustat in rodents, systemic exposures were not continuous throughout the dosing period. As such, possible off-target carcinogenic effects may be underestimated.
In the 2-year mouse carcinogenicity study, significant increases in the incidence of lung bronchoalveolar carcinoma was noted in the low and high dose groups (systemic exposures approximately 1-fold and approximately 3-fold the total AUC at MRHD). A significant increase in subcutis fibrosarcoma was seen in females at the high dose group (systemic exposures approximately 3-fold total AUC at MRHD).
In the 2-year rat carcinogenicity study, a significant increase in the incidence of mammary gland adenoma was noted at the middle dose level (systemic exposure less than 1-fold the total AUC at MRHD). However, the finding was not dose related and the incidence of this tumour type was lower at the highest dose level tested (systemic exposure approximately 2-fold the total AUC at MRHD) and was therefore not considered test article related.
Similar findings from the mouse and rat carcinogenicity studies were not observed in the clinical studies.
Reproductive and developmental toxicity
Roxadustat had no effect on mating or fertility in treated male or female rats at approximately 4-fold the human exposure at the MRHD. However, at the NOAEL in male rats, there were decreases in weights of the epididymis and the seminal vesicles (with fluid) without effects on male fertility. The NOEL for any male reproductive organ related findings was 1.6-fold MRHD. In female rats there were increases in the number of non-viable embryos and post-implantation losses at this dose level compared to control animals.
Results from the reproductive and developmental toxicity studies in rats and rabbits demonstrated reduction of average foetal or pup body weight, average placental weight increase, abortion and pup mortalities.
Pregnant Sprague-Dawley rats administered roxadustat daily from implantation through the closure of the hard palate (Gestation Days 7 – 17) showed decreased foetal body weight and increased skeletal alterations at approximately 6-fold the total AUC at MRHD. Roxadustat had no effect on post-implant foetal survival.
Pregnant New Zealand rabbits were administered roxadustat daily from Gestation Day 7 through Gestation Day 19 and Caesarian sections were performed on Gestation Day 29. Roxadustat administration at systemic exposures up to approximately 3-fold the total AUC at MRHD showed no embryo-foetal findings. However, one doe aborted at approximately 1-fold the total AUC at MRHD and 2 does aborted at approximately 3-fold the total AUC at MRHD, the aborting females showed thin body condition.
In the perinatal/postnatal development study in Sprague-Dawley rats, pregnant dams were administered roxadustat daily from Gestation Day 7 to Lactation Day 20. During the lactation period, pups from dams administered roxadustat at approximately 2-fold the total Cmax at MRHD showed high mortality during the preweaning period and were sacrificed at weaning. Pups from dams administered roxadustat at doses resulting in systemic exposures approximately 3-fold the human exposure at MRHD showed a significant decrease in 21-day survival after birth (lactation index) compared with pups from control litters.
In a cross-fostering study, the most pronounced effects on rat pup viability were noted in the pups exposed to roxadustat postnatally only, and the pup viability exposed to roxadustat until delivery was lower than that of unexposed pups.
The cross-fostering study in which pups from unexposed rats were cross fostered with dams treated with roxadustat (human equivalent dose approximately 2-fold MRHD), had roxadustat in pup plasma indicating transfer of drug via the milk. Milk from these dams had roxadustat present. The pups who were exposed to milk containing roxadustat showed a lower survival rate (85.1%) versus pups from untreated dams cross fostered with untreated dams (98.5% survival rate). The mean body weight of the surviving pups exposed to roxadustat during the lactation period was also less than the control pups (no in utero exposure – no exposure in milk).
Cardiovascular safety
A cardiovascular safety pharmacology study showed heart rate increases following a single administration of 100 mg/kg roxadustat to monkeys. There was no effect on hERG or ECG. Additional safety pharmacology studies in rats have shown that roxadustat reduced total peripheral resistance followed by a reflex increase in heart rate from approximately six times the exposure at the MRHD.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.