EVUSHELD Solution for injection Ref.[49908] Active ingredients: Tixagevimab and Cilgavimab
Source: European Medicines Agency (EU) Revision Year: 2022 Publisher: AstraZeneca AB, SE-151 85 Södertälje, Sweden
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Immune sera and immunoglobulins, antiviral monoclonal antibodies
ATC code: J06BD03
Mechanism of action
Tixagevimab and cilgavimab are two recombinant human IgG1 monoclonal antibodies, with amino acid substitutions in the Fc regions, to extend antibody half-life and to reduce antibody effector function and potential risk of antibody-dependent enhancement of disease (see section 5.3). Tixagevimab and cilgavimab can simultaneously bind to non-overlapping regions of the spike protein receptor binding domain (RBD) of SARS-CoV-2. Tixagevimab, cilgavimab and their combination bind to spike with equilibrium dissociation constants of KD = 2.76 pM, 13.0 pM and 13.7 pM, respectively, blocking its interaction with the human ACE2 receptor, resulting in a blockade of virus entry. Tixagevimab, cilgavimab and their combination blocked RBD binding to the human ACE2 receptor with IC50 values of 0.32 nM (48 ng/mL), 0.53 nM (80 ng/mL) and 0.43 nM (65 ng/mL), respectively.
Antiviral activity
In a SARS-CoV-2 virus neutralisation assay on Vero E6 cells, tixagevimab, cilgavimab and their combination neutralised SARS-CoV-2 (USA-WA1/2020 isolate) with EC50 values of 60.7 pM (9 ng/mL), 211.5 pM (32 ng/mL) and 65.9 pM (10 ng/mL), respectively. These in-vitro values correlate with in-vivo clinically effective serum concentrations of 2.2 µg/mL of EVUSHELD.
Antiviral resistance
SARS-CoV-2 or recombinant vesicular stomatitis virus encoding SARS-CoV-2 spike protein (pseudovirus) were serially passaged in cell cultures in the presence of tixagevimab or cilgavimab individually, or tixagevimab and cilgavimab in combination. Escape variants were identified following passage with cilgavimab, but not with tixagevimab or tixagevimab and cilgavimab in combination.
In neutralisation assays using recombinant SARS-CoV-2 pseudoviruses harbouring individual spike substitutions identified in circulating SARS-CoV-2, variants with reduced susceptibility to tixagevimab alone included those with F486S (>600-fold) and F486V (121- to 149-fold) and variants with reduced susceptibility to cilgavimab alone included those with R346I (>200-fold), K444E (>200-fold), K444Q (>200-fold) and K444R (>200-fold).
Tixagevimab and cilgavimab in combination retained full to nearly full neutralisation activity against pseudovirus and/or live virus SARS-CoV-2 variant strains harbouring all spike substitutions identified in Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1) Delta (B.1.617.2) and Delta [+K417N] (AY.1/AY.2), and Omicron (BA.2) variants of concern. Pseudotyped VLPs expressing spike protein and authentic SARS-CoV-2 Omicron BA.1 variant (B.1.1.529) and Omicron BA.1.1 (B.1.1.529 [+R346K]) showed reduced susceptibility to tixagevimab and cilgavimab in combination (Table 3).
Data collection is ongoing to better understand how small reductions in activity seen in authentic SARS-CoV-2 or pseudotyped VLP assays may correlate with clinical outcomes.
Table 3. Pseudovirus and authentic SARS-CoV-2 neutralisation data for SARS-CoV-2 variant substitutions with tixagevimab and cilgavimab together:
| Pango Lineage with Spike Protein Substitutions | Characteristic RBD Substitutions Tested | Fold Reduction in Susceptibilitya | IC50 (ng/mL) | ||
|---|---|---|---|---|---|
| Pseudovirusb | Live Virusc | Pseudovirusb | Live Virusc | ||
| Variants of Concern | |||||
| B.1.1.7 (Alpha, UK) | N501Y | 1.3-4.2 | 0.5-1.4 | 2.7-9.0 | 4-39.5 |
| B.1.351 (Beta, South Africa) | K417N:E484K:N501Y | 2.5-5.5 | 0.9-3.8 | 5.6-11.4 | 6.5-256 |
| P.1 (Gamma, Brazil) | K417T:E484K:N501Y | 0.8-1.7 | 0.4-2.0 | 1.8-2.7 | 3.2-8 |
| B.1.617.2 (Delta, India) | L452R:T478K | 1-1.2 | 0.6-1.0 | 1.9-2.2 | 3-7.5 |
| AY.1/AY.2 (Delta [+K417N], India) | K417N:L452R:T478K | 1.0 | ND | 1.9 | ND |
| B.1.1.529 Omicron, BA.1 (Botswana) | G339D:S371L:S373P: S375F:K417N:N440K: G446S:S477N:T478K: E484A:Q493R:G496S: Q489R:N501Y:Y505H | 132-183 | 12-30 | 51-277 | 147-278 |
| Omicron BA.1.1 (Multiple Country origin) | G339D:R346K:S371L: S373P: S375F:K417N: N440K:G446S:S477N: T478K:E484A:Q493R: G496S:Q489R:N501Y: Y505H | 424 | 176 | 466 | 1147 |
| Omicron BA.2 (Multiple Country Origin) | G339D:S371F:S373P: S375F:T376A:D405N: R408S:K417N:N440K: S477N: T478K:E484A: Q493R:Q498R:N501Y: Y505H:H655Y:N679K: P681H:N764K | 3.2 | 5.4 | 9.8 | 35 |
| Variants of Interest | |||||
| B.1.525 (Eta, Multiple country) | E484K | 1.8-3.0 | ND | 5-9.5 | ND |
| B.1.526 (Iota, United States) | E484K | 0.7-3.4 | 0.3-1.8 | 1.8-4.5 | 1.0-7.0 |
| B.1.617.1 (Kappa, India) | L452R:E484Q | 0.9-3.4 | 0.5-1.3 | 2.5-5.1 | 2.0-5.0 |
| C.37 (Lambda, Peru) | L452Q:F490S | 0.7 | ND | 1.1 | ND |
| B.1.621 (Mu, Colombia) | R346K:E484K:N501Y | 7.5 | ND | 13.5 | ND |
| Variant Alerts for Further Monitoring | |||||
| B.1.427 / B.1.429 (Epsilon, United States) | L452R | 0.8-2.9 | 1.3-3.5 | 1.2-4.4 | 5.0-14.0 |
| R.1 (Multiple country) | E484K | 3.5 | ND | 4.6 | ND |
| B.1.1.519 (Multiple country) | T478K | 1.0 | ND | 2.3 | ND |
| C.36.3 (Multiple country) | R346S:L452R | 2.3 | ND | 3.9 | ND |
| B.1.214.2 (Multiple country) | Q414K:N450K | 0.8 | ND | 1.6 | ND |
| B.1.619.1 (Multiple country) | N440K:E484K | 3.3 | ND | 7.6 | ND |
| Variants De-Escalated from Further Monitoring | |||||
| P.2 (Zeta, Brazil) | E484K | 2.9 | ND | 10.4 | ND |
| B.1.616 (France) | V483A | 0.4-0.5 | ND | 1.1-1.2 | ND |
| A.23.1 (UK) | V367F | 0.4 | ND | 0.5 | ND |
| A.27 (Multiple country) | L452R:N501Y | 0.8 | ND | 1.8 | ND |
| AV.1 (Multiple country) | N439K:E484K | 5.9 | ND | 13.0 | ND |
a Range of reduced in-vitro potency across multiple sets of co-occurring substitutions and/or testing labs using research-grade assays; mean fold change in half maximal inhibitory concentration (IC50) of monoclonal antibody required for a 50% reduction in infection compared to wild type reference strain.
b Pseudoviruses expressing the entire SARS-CoV-2 spike variant protein and individual characteristic spike substitutions except L452Q were tested including Alpha (+L455F, E484K, F490S, Q493R, and/or S494P), and Delta (+K417N) harbouring additional indicated RBD substitutions that are no longer detected or detected at extremely low levels within these lineages.
c Authentic SARS-CoV-2 expressing the entire variant spike protein were tested including Alpha (+E484K or S494P) harbouring additional indicated RBD substitutions that are no longer detected or detected at extremely low levels within these lineages.
ND, not determined; RBD, receptor binding domain.
It is not known how pseudovirus or authentic SARS-CoV-2 neutralisation susceptibility data correlate with clinical outcome.
In PROVENT, sequencing data collected at illness visits was available for 21 participants with COVID-19 infection (6 who received tixagevimab and cilgavimab and 15 placebo). At an allele fraction ≥25%, 14 participants were infected with variants of concern or variants of interest, including 8 participants with Alpha (B.1.1.7) (8 placebo), 1 participant with Beta (B.1.351) (1 who received tixagevimab and cilgavimab), 3 participants with Delta (B.1.617.2) (3 placebo), and 2 participants with Epsilon (B.1.429) (2 who received tixagevimab and cilgavimab). Additional spike protein RBD substitutions detected at an allele fraction ≥3% included V503F in the tixagevimab and cilgavimab group.
It is possible that resistance-associated variants to tixagevimab and cilgavimab together could have cross-resistance to other monoclonal antibodies targeting the RBD of SARS-CoV-2. Tixagevimab and cilgavimab together retained activity against pseudoviruses harbouring individual SARS-CoV-2 spike substitutions (E484D/K/Q, F490S, Q493R, S494P, K417E/N, D420N, K444Q, V445A, Y453F, L455F, N460K/S/T, F486V, and Q493K) identified in neutralisation escape variants of other monoclonal antibodies targeting the RBD of SARS-CoV-2 spike protein.
Pharmacodynamic effects
In PROVENT, following an intramuscular dose of 150 mg tixagevimab and 150 mg cilgavimab, neutralising antibody GMT at 7 (n=891), 28 (n=954) and 57 (n=43) days post-dose were similar to those observed in the Phase I healthy volunteer study and were 16-, 22-, and 17-fold higher, respectively, than the GMT measured in convalescent plasma from COVID-19 patients (GMT=30.8).
Immunogenicity
In PROVENT through Day 183, treatment-emergent anti-tixagevimab, anti-cilgavimab and antiEVUSHELD antibodies were detected in 0.8% (6/716), 1.1% (7/644) and 1.3% (10/743) ADAevaluable participants who received EVUSHELD. No evidence of an association of ADA with any impact on efficacy or safety has been observed.
Clinical efficacy
PROVENT
PROVENT is an ongoing Phase III, randomised (2:1), double-blind, placebo-controlled clinical trial studying EVUSHELD for the pre-exposure prophylaxis of COVID-19 in adults ≥18 years of age. Enrolled participants were individuals considered to be at increased risk for inadequate response to active immunisation (due to age ≥60 years, co-morbidity, pre-existing chronic illness, immunocompromised, or intolerant of vaccination) or at increased risk of SARS-CoV-2 infection (due to their location or circumstances at time of enrolment, for example health care workers including staff for long-term care facilities, working in high risk industrial settings or living with high density proximity, including students in dormitories and military barracks). Participants received either 150 mg of tixagevimab and 150 mg of cilgavimab or placebo, administered as two separate intramuscular injections. The study excluded participants with a history of laboratory-confirmed SARS-CoV-2 infection or SARS-CoV-2 antibody positivity at screening.
The baseline demographics were well balanced across the EVUSHELD and placebo arms. The median age was 57 years (with 24% of participants aged 65 years or older and 4% of participants aged 75 years or older), 46% of participants were female, 73% were White, 3% were Asian, 17% were Black/African American, and 15% were Hispanic/Latino. Of the 5 197 participants, 78% had baseline co-morbidities or characteristics associated with an increased risk for severe COVID-19, including obesity (42%), diabetes (14%), cardiovascular disease (8%), cancer, including a history of cancer (7%), chronic obstructive pulmonary disease (5%), chronic kidney disease (5%), chronic liver disease (5%), immunosuppressive medications (3%) and immunosuppressive disease (<1%).
The primary analysis included 5 172 participants who were SARS-CoV-2 RT-PCR-negative at baseline, of which 3 441 received EVUSHELD and 1 731 received placebo. EVUSHELD significantly (p-value<0.001) reduced the risk of SARS-CoV-2 RT-PCR-positive symptomatic illness (COVID-19) when compared to placebo (Table 4). The median follow-up time post-administration was 83 days.
Table 4. Incidence of COVID-19:
| N | Number of eventsa, n (%) | Relative Risk Reduction, % (95% CI) | |
|---|---|---|---|
| EVUSHELDb | 3,441 | 8 (0.2%) | 77% (46-90) |
| Placebo | 1,731 | 17 (1.0%) |
CI = Confidence Interval, N = number of participants in analysis.
a Primary endpoint, a participant was defined as a COVID-19 case if their first case of SARS-CoV-2 RT-PCR-positive symptomatic illness occurred after administration and prior to Day 183.
b 150 mg tixagevimab and 150 mg cilgavimab.
Efficacy was consistent across pre-defined sub-groups including age, gender, ethnicity and baseline co-morbidities or characteristics associated with an increased risk for severe COVID-19.
Among participants who received EVUSHELD there were no severe/critical COVID-19 events (defined as SARS-CoV-2 RT-PCR-positive symptomatic illness characterised by a minimum of either pneumonia [fever, cough, tachypnoea or dyspnoea, and lung infiltrates] or hypoxemia [SpO2 <90% in room air and/or severe respiratory distress] and a WHO Clinical Progression Scale score of 5 or higher) compared to one event (0.1%) among participants who received placebo.
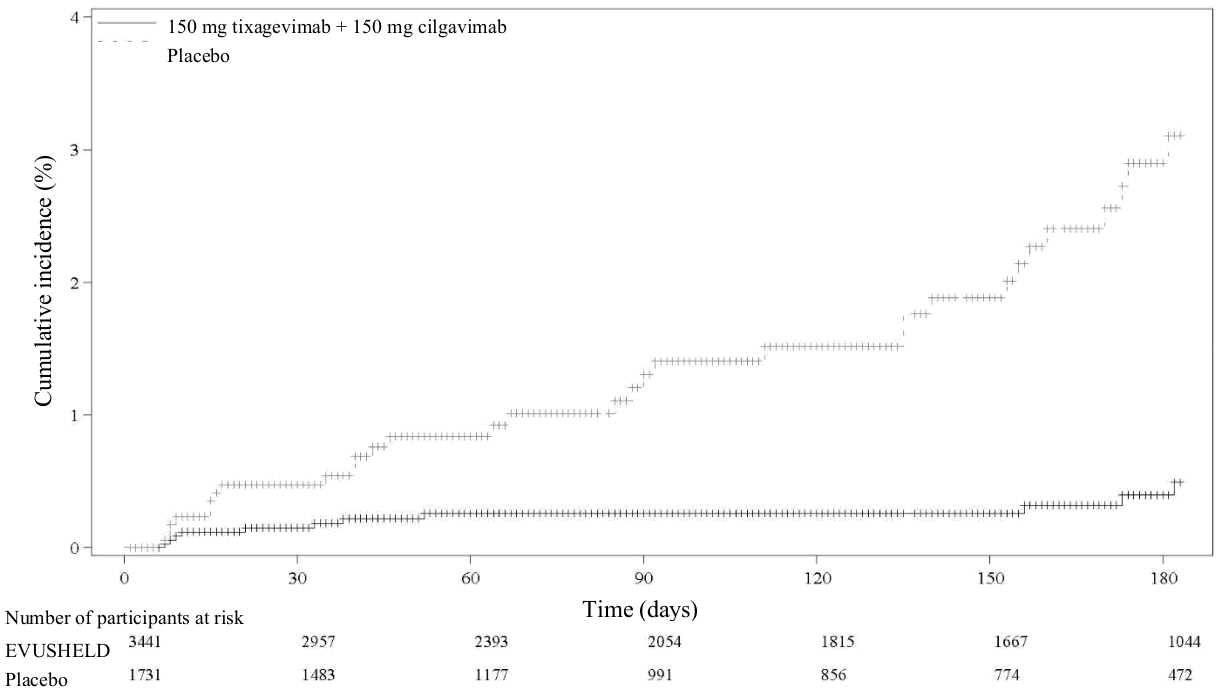
An additional data cut-off was conducted to provide post-hoc updated safety and efficacy analyses; the median follow-up was 6.5 months for participants in both the EVUSHELD and placebo arms. The relative risk reduction of SARS-CoV-2 RT-PCR-positive symptomatic illness was 83% (95% CI 66-91), with 11/3 441 (0.3%) events in the EVUSHELD arm and 31/1 731 (1.8%) events in the placebo arm, see Figure 1). Among participants who received EVUSHELD there were no severe/critical COVID-19 events compared to five events among participants who received placebo.
In exploratory analyses of all participants who received EVUSHELD or placebo, including 25 participants who were subsequently found to have been SARS-CoV-2 RT-PCR-positive at baseline, the relative risk reduction of SARS-CoV-2 RT-PCR-positive symptomatic illness was 78% (95% CI 59-88), with 14/3 460 (0.4%) events in the EVUSHELD arm and 31/1 737 (1.8%) events in the placebo arm at a median follow-up of 6.5 months.
Figure 1. Kaplan Meier: Cumulative Incidence of Symptomatic COVID-19:
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with EVUSHELD in one or more subsets of the paediatric population in the prophylaxis of COVID-19 (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
The pharmacokinetics of tixagevimab and cilgavimab are comparable, linear and dose-proportional between 150 mg tixagevimab and 150 mg cilgavimab and 1,500 mg of tixagevimab and 1,500 mg of cilgavimab following a single intravenous administration.
Absorption
After an intramuscular dose of 150 mg tixagevimab and 150 mg cilgavimab in healthy volunteers, the mean (% CV) maximum concentration (Cmax) was 16.5 (35.6%) and 15.3 (38.5%) µg/mL for tixagevimab and cilgavimab, respectively, which was reached at a median Tmax of 14 days. The estimated absolute bioavailability after a single 150 mg intramuscular dose was 68.5% for tixagevimab and 65.8% for cilgavimab.
Based on pharmacokinetic/pharmacodynamic modelling, the time to achieve the minimum protective serum concentration (2.2 µg/mL) is estimated to be 6 hours following intramuscular administration of 150 mg tixagevimab and 150 mg cilgavimab into the gluteal region.
Distribution
Based on PK modelling, the central volume of distribution was 2.72 L for tixagevimab and 2.48 L for cilgavimab. The peripheral volume of distribution was 2.64 L for tixagevimab and 2.57 L for cilgavimab.
Biotransformation
Tixagevimab and cilgavimab are expected to be degraded into small peptides and component amino acids via catabolic pathways in the same manner as endogenous IgG antibodies.
Elimination
The clearance (CL) was 0.041 L/day for tixagevimab and 0.041 L/day for cilgavimab with interindividual variability of 21% and 29% respectively. The estimated population median terminal elimination half-life was 89 days for tixagevimab and 84 days for cilgavimab.
In PROVENT, following a single intramuscular dose of 150 mg tixagevimab and 150 mg cilgavimab, the median EVUSHELD serum concentration was 8.3 µg/mL (range 1.3 to 19.5 µg/mL) on Day 183.
Special populations
Renal impairment
No specific studies have been conducted to examine the effects of renal impairment on the pharmacokinetics of tixagevimab and cilgavimab.
Tixagevimab and cilgavimab are not eliminated intact in the urine, thus renal impairment is not expected to significantly affect the exposure of tixagevimab and cilgavimab. Similarly, dialysis is not expected to impact the PK of tixagevimab and cilgavimab.
Based on population PK analysis, there is no difference in the clearance of tixagevimab and cilgavimab in patients with mild (N=978) or moderate (N=174) renal impairment compared to patients with normal renal function. In the population PK model there were insufficient participants with severe renal impairment (N=21) to draw conclusions.
Hepatic impairment
No specific studies have been conducted to examine the effects of hepatic impairment on the PK of tixagevimab and cilgavimab. The impact of hepatic impairment on the PK of tixagevimab and cilgavimab is expected to be low.
Tixagevimab and cilgavimab are expected to be catabolised by multiple tissues through proteolytic degradation into amino acids and recycling into other proteins, therefore hepatic impairment is not expected to affect the exposure of tixagevimab and cilgavimab.
Elderly
Of the 2 560 participants in the pooled PK analysis, 21% (N=534) were 65 years of age or older and 4.2% (N=107) were 75 years of age or older. There is no clinically meaningful difference in the PK of tixagevimab and cilgavimab in geriatric subjects (≥65 years) compared to younger individuals.
Paediatric population
The PK of tixagevimab and cilgavimab in individuals <18 years old has not been evaluated.
Using population PK modelling and simulation, the recommended dosing regimen is expected to result in comparable serum exposures of tixagevimab and cilgavimab in adolescents aged 12 years or older who weigh at least 40 kg as observed in adults, since adults with similar body weight have been included in the clinical trial PROVENT.
High body weight
Based on population PK analysis, a decrease in EVUSHELD serum concentrations was observed with increased body weight. The average serum concentration in an adult weighing >95 kg following a 150 mg tixagevimab and 150 mg cilgavimab intramuscular administration was predicted to be approximately 37% lower than in an adult weighing 65 kg.
Other special populations
Based on a population PK analysis, sex, age, race, ethnicity, cardiovascular disease, diabetes and immunocompromise had no clinically relevant effect on the PK of tixagevimab and cilgavimab.
5.3. Preclinical safety data
Carcinogenesis, mutagenesis, and reproductive toxicology studies have not been conducted with tixagevimab and cilgavimab.
Non-clinical data reveal no special hazard for humans based on studies of tissue binding and a singledose toxicity study in cynomolgus monkeys including assessment of safety pharmacology and local tolerance.
Antibody-dependent enhancement (ADE) of infection
The potential of tixagevimab and cilgavimab to mediate antibody-dependent viral entry was assessed in FcγRII-expressing Raji cells co-incubated with recombinant virus pseudotyped with SARS-CoV-2 spike protein, with antibody concentrations at a range of 6.6 nM (1 µg/mL) to 824 pM (125 ng/mL). Tixagevimab, cilgavimab and their combination did not mediate entry of pseudovirus into these cells.
The potential for ADE was also evaluated in a non-human primate model of SARS-CoV-2 using EVUSHELD. Intravascular administration prior to virus inoculation resulted in a dose-dependent improvement in all measured outcomes (total viral RNA in the lungs or nasal mucosae, infectious virus levels in the lungs based on TCID50 measurements, and lung injury and pathology based on histology measurements). No evidence of enhancement of disease was observed at any dose evaluated, including sub-neutralizing doses down to 0.04 mg/kg.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.