EXDENSUR Solution for injection Ref.[116332] Active ingredients: Depemokimab
Source: European Medicines Agency (EU) Revision Year: 2026 Publisher: GlaxoSmithKline Trading Services Limited, 12 Riverwalk, Citywest Business Campus, Dublin 24, D24 YK11, Ireland
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Drugs for obstructive airway diseases, other systemic drugs for obstructive airways diseases
ATC code: R03DX12
Mechanism of action
Depemokimab targets human IL-5 with a binding affinity of 10.5 pM, thereby blocking the binding to the IL-5 receptor alpha expressed on the cell surface with picomolar potency (IC50 4 pM) in vitro. Depemokimab contains a triple amino acid substitution (YTE) in the fragment crystallisable (Fc) region which increases binding to the neonatal Fc receptor and thereby extends the half-life when compared to the IgG1 wildtype.
IL-5 is a pleiotropic cytokine with established impact on eosinophils and other immune and structural cells. In severe asthma, inhibition of IL-5 has demonstrated an improvement in epithelial integrity, mucus plugging and reduction in tissue remodelling. However, the mechanism of action has not been definitively established.
Pharmacodynamic effects
In a clinical pharmacology study with mild-to-moderate asthma patients, a single 100 mg subcutaneous dose of depemokimab produced a rapid reduction in blood eosinophil count. Blood eosinophils were reduced by 54% compared to placebo 24 hours after dosing, which was the first post-dose assessment.
In the asthma and CRSwNP phase 3 studies, these reductions were sustained over the treatment period and blood eosinophil count reductions at week 52 were 79% and 85% compared to placebo, respectively.
Immunogenicity
In patients who received at least one 100 mg dose of depemokimab administered subcutaneously every 6 months, 9% (44/499) of patients with asthma (SWIFT-1 and SWIFT-2) and 8% (21/272) of patients with CRSwNP (ANCHOR-1 and ANCHOR-2) were positive for anti-depemokimab antibodies (ADA) during the 52-week studies.
The percentage of patients who were positive for ADA was 9% (55/622) in a 52-week open-label extension asthma study (AGILE; n=395 with data collected for 104 weeks).
Across the placebo-controlled studies for asthma and CRSwNP indications, and the 52-week open label extension asthma study (AGILE), <1% of the patients (n=7) were positive for neutralising antibodies.
Anti-drug antibodies (ADA) were commonly detected. No evidence of ADA impact on pharmacokinetics, pharmacodynamics, efficacy or safety was observed; however, data are still limited.
Clinical efficacy and safety
Asthma
The efficacy of depemokimab was evaluated in 2 replicate, randomised (2:1 ratio, depemokimab to placebo), double-blind, placebo-controlled, parallel-group, multi-centre clinical studies of 52-weeks treatment duration (SWIFT-1 and SWIFT-2). The two studies enrolled patients aged 12 years and older with asthma with type 2 inflammation characterised by an eosinophilic phenotype. In these studies, depemokimab 100 mg was administered subcutaneously once every 6 months for a total of 2 doses in addition to standard of care (SoC) therapy. Patients were required to have 2 or more asthma exacerbations requiring treatment with systemic corticosteroids (SCS) in the last 12 months, while on medium- to high-dose ICS (≥440 mcg fluticasone propionate or equivalent) plus at least one additional asthma controller. Patients were also required to have a blood eosinophil count of >150 cells/mcL at screening or >300 cells/mcL documented in the year prior to study entry and reduced lung function at baseline (pre-bronchodilator forced expiratory volume in 1 second [FEV1] <80% predicted normal in adults and [FEV1] <90% or FEV1:FVC ratio <0.8 in adolescents). Patients were enrolled without requiring a minimum baseline Asthma Control Questionnaire-5 (ACQ-5) score. Depemokimab was administered as add-on to background asthma treatment which continued throughout the duration of the studies. The Full Analysis Set (FAS) population consisted of 762 patients who were randomised and received at least one dose of depemokimab or placebo in the two studies (382 in SWIFT-1 and 380 in SWIFT-2).
The demographics and baseline characteristics of the patients in these 2 studies are provided in Table 2.
Table 2. Demographics and baseline characteristics (FAS population):
| SWIFT-1 (N=382) | SWIFT-2 (N=380) | |
|---|---|---|
| Age (y) of patients, mean (SD) | 54 (14.2) | 53 (16.2) |
| Patients aged ≥65 years, n (%) | 98 (26) | 96 (25) |
| Female, n (%) | 223 (58) | 241 (63) |
| White, n (%) | 316 (83) | 272 (72) |
| Duration of asthma, years, mean (SD) | 22 (16.2) | 25 (18.5) |
| Mean pre-bronchodilator % predicted FEV1 (SD) | 62 (15.2) | 62 (15.9) |
| Mean % reversibility (SD) | 17 (15.3) | 18 (17.4) |
| Mean number of exacerbations in previous year (SD) | 2.2 (0.69) | 2.7 (1.92) |
| Eosinophil count, cells/mcL, median (min, max) | 310 (20, 2 360) | 340 (10, 4 440) |
| Total IgE, U/mcL, median (min, max) | 185 (1.9, 12 142) | 180 (2.2, 16 198) |
| Mean SGRQ total score (SD), range 0-100 | 44.3 (20.70) | 44.5 (18.69) |
| Patients with ACQ ≥1.5 at baseline, n (%) | 280 (75) | 279 (75) |
| Medium-dose ICS use, n (%)a | 179 (47) | 154 (41) |
| High-dose ICS use, n (%)a | 203 (53) | 226 (59) |
| ICS + LAMA + LABA use, n (%) | 95 (25) | 127 (33) |
| Maintenance OCS use, n (%) | 21 (5) | 19 (5) |
FAS = Full Analysis Set, FEV1 = Forced Expiratory Volume in 1 second, IgE = immunoglobulin E, SGRQ = St. George's Respiratory Questionnaire, ACQ-5 = Asthma Control Questionnaire, ICS = inhaled corticosteroid, OCS = oral corticosteroid
a Medium-dose ICS = 440 mcg FP daily or equivalent; High-dose ICS >440 mcg FP daily or equivalent
Exacerbations
The primary efficacy endpoint for SWIFT-1 and SWIFT-2 was the annualised rate of clinically significant exacerbations over the 52-week treatment period. A clinically significant exacerbation was defined as worsening of asthma requiring use of SCS (intravenous or oral steroids for at least 3 days or a single intramuscular corticosteroid dose) and/or hospitalisation and/or emergency department visit. For patients on maintenance SCS, at least double the existing maintenance dose for at least 3 days was required. All patients experiencing an exacerbation were treated with SCS. The majority of patients (95% and 92% for SWIFT-1 and SWIFT-2, respectively) completed the studies.
In SWIFT-1 and SWIFT-2, the annualised rate of asthma exacerbations was significantly lower in patients receiving depemokimab compared to placebo (Table 3). The percentage of patients with exacerbations requiring hospitalisation and/or emergency department visit was lower for patients treated with depemokimab (1% and 4%) compared with placebo (8% and 10%) in SWIFT-1 and SWIFT-2, respectively.
Table 3. Results of primary exacerbation endpoint (FAS population):
| SWIFT-1 | SWIFT-2 | |||
|---|---|---|---|---|
| Depemokimab N=250 | Placebo N=132 | Depemokimab N=252 | Placebo N=128 | |
| Annualised asthma exacerbations rate | ||||
| Percent of patients with an exacerbation | 32% | 46% | 32% | 50% |
| Exacerbation rate per year | 0.46 | 1.11 | 0.56 | 1.08 |
| Rate ratio (95% CI) Percent reduction (95% CI) | 0.42 (0.30, 0.59) 58% (41, 70) | 0.52 (0.36, 0.73) 48% (27, 64) | ||
| p-value | <0.001 | <0.001 | ||
FAS = Full Analysis Set
Secondary endpoints
Additional efficacy assessments included health-related quality of life measured with St. George's Respiratory Questionnaire (SGRQ), asthma control measured with Asthma Control Questionnaire (ACQ-5) and lung function (pre-bronchodilator FEV1). Table 4 provides the results of these secondary endpoints for the FAS population of SWIFT-1 and SWIFT-2.
Table 4. Results of secondary endpoints (FAS population):
| SWIFT-1 | SWIFT-2 | |||
|---|---|---|---|---|
| Depemokimab N=250 | Placebo N=132 | Depemokimab N=252 | Placebo N=128 | |
| St. George's Respiratory Questionnaire (SGRQ) total score at Week 52 | ||||
| na | 224 | 114 | 224 | 116 |
| LS mean change from baseline (SE) | -13.0 (1.11) | -9.7 (1.55) | -14.8 (1.04) | -12.5 (1.46) |
| Adjusted treatment differenceb | -3.4 | -2.3 | ||
| (95% CI) | (-7.1, 0.4) | (-5.8, 1.2) | ||
| Asthma Control Questionnaire-5 (ACQ-5) score at Week 52 | ||||
| na | 224 | 114 | 224 | 116 |
| LS mean change from baseline (SE) | -0.82 (0.066) | -0.77 (0.091) | -0.81 (0.065) | -0.70 (0.091) |
| Adjusted treatment differenceb | -0.04 | -0.11 | ||
| (95% CI) | (-0.27, 0.18) | (-0.33, 0.11) | ||
| Pre-bronchodilator FEV1 (mL) at Week 52 | ||||
| na | 224 | 115 | 226 | 112 |
| LS mean change from baseline (SE) | 160 (26.3) | 160 (36.4) | 240 (28.6) | 184 (40.7) |
| Adjusted treatment differenceb | -1 | 56 | ||
| (95% CI) | (-89, 88) | (-43, 154) | ||
FAS = Full Analysis Set, LS = Least Squares, FEV1 = Forced Expiratory Volume in 1 second
a Number of patients with analysable data at the timepoint
b Adjusted treatment difference (depemokimab vs. placebo)
Open-label extension study in asthma (AGILE)
Patients who completed either the SWIFT-1 or SWIFT-2 study were able to enrol in an open-label extension study (AGILE) where they all received up to two doses of depemokimab over an additional 52 weeks. The analysis of AGILE (n=629) showed an annualised exacerbation rate of 0.56 (95% CI: 0.49, 0.65).
Chronic rhinosinusitis with nasal polyps (CRSwNP)
The efficacy of depemokimab in adult patients with CRSwNP was evaluated in 2 replicate, randomised, double-blind, placebo-controlled, parallel-group, multicentre clinical studies of 52-weeks duration (ANCHOR-1 and ANCHOR-2). These studies evaluated the efficacy of 100 mg of depemokimab administered subcutaneously once every 6 months for a total of 2 doses in addition to standard of care (SoC) therapy. Patients had been treated with systemic corticosteroids (SCS) anytime within the past 2 years; and/or had a medical contraindication/intolerance to SCS; and/or had a documented history of prior surgery for nasal polyps (NP) prior to screening. Randomised patients were required to have an endoscopic bilateral NP score of at least 5 out of a maximum score of 8 with a minimum score of 2 in each nasal cavity and a mean nasal obstruction Verbal Response Scale (VRS) score of 2 or greater at baseline. Other than nasal obstruction, there were no other entry requirements for symptoms or for quality of life at randomisation. A total of 528 patients (271 in ANCHOR-1 and 257 in ANCHOR-2) were included in the Full Analysis Set (FAS) population.
The demographics and baseline characteristics of the patients in these two studies are provided in Table 5 below:
Table 5. Demographics and baseline characteristics (FAS population):
| ANCHOR-1 N=271 | ANCHOR-2 N=257 | |
|---|---|---|
| Age (y) of patients, mean (SD) | 54 (13.4) | 50 (12.9) |
| Patients aged ≥65 years, n (%) | 57 (21) | 43 (17) |
| Female, n (%) | 83 (31) | 80 (31) |
| White, n (%) | 185 (70) | 197 (77) |
| Duration (y) of CRSwNP, mean (SD) | 13 (11.2) | 11 (8.7) |
| Blood eosinophil count, cells/mcL, median (min, max) | 360 (10, 10 550) | 360 (30, 1 670) |
| Intranasal corticosteroid use, n (%) | 265 (98) | 249 (97) |
| Patients with ≥1 previous NP surgery, n (%) | 171 (63) | 162 (63) |
| SCS use for NP in past 12 months, n (%) | 190 (70) | 172 (67) |
| Medical contraindication/intolerance to SCS, n (%) | 11 (4) | 13 (5) |
| Asthma, n (%) | 161 (59) | 131 (51) |
| AERD, n (%) | 43 (16) | 42 (16) |
| Total endoscopic NP scoreabc, mean (SD), maximum score = 8 | 6.0 (1.35) | 5.9 (1.29) |
| Nasal obstruction VRS mean scoread, mean (SD), maximum score = 3 | 2.5 (0.48) | 2.6 (0.42) |
| Loss of smell VRS mean scoread, mean (SD), maximum score = 3 | 2.7 (0.55) | 2.8 (0.41) |
| SNOT-22 total scoreae, mean (SD), maximum score = 110 | 57.4 (22.15) | 60.1 (19.95) |
| Patients with SNOT-22 total score ≥40, n (%) | 204 (75) | 207 (81) |
FAS = Full Analysis Set, CRSwNP = chronic rhinosinusitis with nasal polyps, SCS = systemic corticosteroid, NP = nasal polyp, AERD = aspirin-exacerbated respiratory disease, VRS = Verbal Response Scale, SNOT-22=Sino-Nasal Outcome Test
a Higher scores indicate greater disease severity.
b As graded by independent blinded assessors.
c NP score is the sum of scores from both nostrils (0-8 scale) where each nostril was graded (0=no polyps; 1=small polyps in the middle meatus not reaching below the inferior border of the middle concha; 2=polyps reaching below the lower border of the middle turbinate; 3=large polyps reaching the lower border of the inferior turbinate or polyps medial to the middle concha; 4=large polyps causing almost complete congestion/obstruction of the inferior meatus).
d Collected daily by patients on a 0 to 3 scale where 0=no symptoms, 1=mild symptoms, 2=moderate symptoms, 3=severe symptoms.
e SNOT-22 is a health-related quality of life assessment tool and includes 22 items in 6 domains of symptoms and impact associated with CRSwNP (nasal, non-nasal, ear/facial, sleep, fatigue, emotional consequences). Higher scores indicate worse health related quality of life.
Total endoscopic nasal polyp score and nasal obstruction VRS score
The co-primary efficacy endpoints in each study were change from baseline in total endoscopic NP score (0-8 scale) at Week 52 as graded by central blinded readers and change from baseline in mean nasal obstruction VRS score (0-3 scale [0=no symptoms, 1=mild symptoms, 2=moderate symptoms, 3=severe symptoms]) over Weeks 49 to 52 as self-reported by patients using a daily diary. The results for the co-primary endpoints in the ANCHOR-1 and ANCHOR-2 studies are presented in Table 6.
Table 6. Results of co-primary endpoints (FAS population):
| ANCHOR-1 | ANCHOR-2 | |||
|---|---|---|---|---|
| Depemokimab N=143 | Placebo N=128 | Depemokimab N=129 | Placebo N=128 | |
| Total endoscopic NP score at Week 52ab | ||||
| nc | 128 | 120 | 120 | 115 |
| LS mean (SE) | 5.4 (0.14) | 6.2 (0.15) | 5.4 (0.14) | 6.0 (0.15) |
| LS mean change from baseline (SE) | -0.6 (0.14) | 0.2 (0.15) | -0.5 (0.14) | 0.1 (0.15) |
| Adjusted treatment differenced (95% CI) | -0.7 (-1.1, -0.3) | -0.6 (-1.0, -0.2) | ||
| p-value | <0.001 | 0.004 | ||
| Nasal obstruction VRS mean score over Weeks 49 to 52ab | ||||
| nc | 125 | 116 | 119 | 111 |
| LS mean (SE) | 1.77 (0.079) | 2.00 (0.083) | 1.83 (0.076) | 2.07 (0.078) |
| LS mean change from baseline (SE) | -0.76 (0.079) | -0.53 (0.083) | -0.77 (0.076) | -0.53 (0.078) |
| Adjusted treatment differenced (95% CI) | -0.23 (-0.46, 0.00e) | -0.25 (-0.46, -0.03) | ||
| p-value | 0.047 | 0.025 | ||
FAS = Full Analysis Set, NP = nasal polyp, LS = Least Squares, VRS = Verbal Response Scale
a Patients who had nasal surgery or used other maintenance treatment impacting type 2 inflammation (including biologics indicated for CRSwNP, chronic use of systemic corticosteroids and intranasal corticosteroids) prior to the timepoint of interest were assigned the worst possible value of the relevant score for all assessments following surgery or initiation of other maintenance treatment impacting type 2 inflammation.
b Based on Mixed Model Repeat Measures (MMRM) analyses with covariates of treatment, baseline score, log(e) baseline blood eosinophil count, region, previous surgery for nasal polyps, visit and interaction terms for visit by baseline and visit by treatment.
c Number of patients with analysable data at the timepoint
d Adjusted treatment difference (depemokimab vs. placebo)
e The upper limit of the 95% CI represents a rounded number of -0.003.
In analyses from the individual studies ANCHOR-1 and ANCHOR-2, a treatment difference in favour of depemokimab was observed by Week 12 (first timepoint assessed) for the total endoscopic NP score and by Weeks 1-4 (first timepoint assessed) for the nasal obstruction VRS mean score that was maintained up to Week 52 (Figures 1 and 2).
Figure 1. LS mean change from baseline (95% CI) in total endoscopic NP score up to Week 52 from ANCHOR-1 and ANCHOR-2 studies (FAS population):
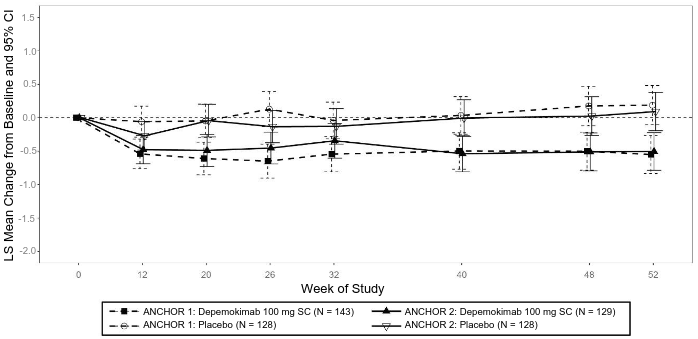
LS = Least Squares; NP = nasal polyp; FAS = Full Analysis Set
Figure 2. LS mean change from baseline (95% CI) in nasal obstruction VRS mean score up to Weeks 49-52 from ANCHOR-1 and ANCHOR-2 studies (FAS population):
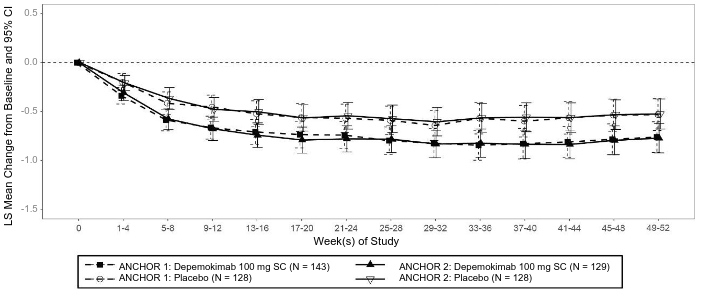
LS = Least Squares; VRS = Verbal Response Scale; FAS = Full Analysis Set
Nasal surgery, systemic corticosteroid use, initiation of other maintenance treatment impacting type 2 inflammation for CRSwNP
Across the two ANCHOR studies, the key secondary endpoint of the proportion of patients who required nasal surgery (actual or planned) or initiated other maintenance treatment impacting type 2 inflammation (including biologics indicated for CRSwNP, chronic use of systemic corticosteroids and intranasal corticosteroids) was 16% (44/272) in the depemokimab group and 22% (56/256) in the placebo group (27% risk reduction; HR: 0.735; 95% CI: 0.495, 1.092, Figure 3). The proportion of patients who had nasal surgery or initiated other maintenance treatment impacting type 2 inflammation for CRSwNP was 12% (33/272) in the depemokimab group, compared with 17% (43/256) in the placebo group, representing a 29% risk reduction (HR: 0.713; 95% CI: 0.453, 1.124).
Figure 3. Kaplan Meier curve for time to first nasal polyps surgery (actual or planned) or initiation of other maintenance treatment impacting type 2 inflammation1 for CRSwNP up to Week 52 (pooled FAS population):
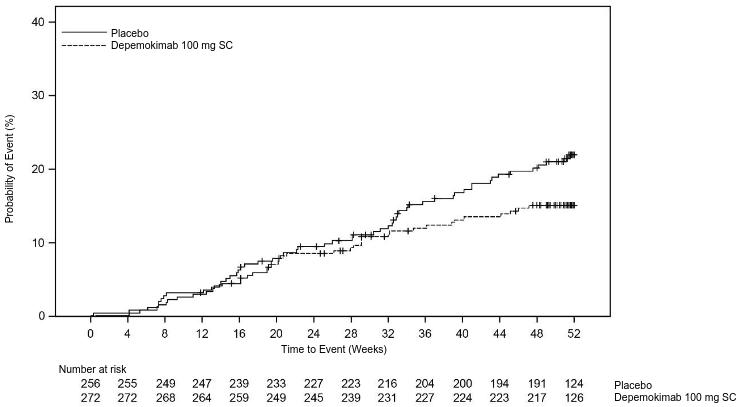
CRSwNP = chronic rhinosinusitis with nasal polyps; FAS = Full Analysis Set
1 Other maintenance treatment impacting type 2 inflammation includes biologics indicated for CRSwNP, chronic use of systemic corticosteroids and intranasal corticosteroids.
Across the two ANCHOR studies, the proportion of patients that required at least 1 course of SCS for CRSwNP or other maintenance treatment impacting type 2 inflammation for CRSwNP or nasal surgery was 26% (72/272) in the depemokimab group compared with 36% (92/256) in the placebo group (OR: 0.58, 95% CI: 0.40, 0.86).
Paediatric population
Asthma
In the SWIFT-1 and SWIFT-2 studies, there were 30 adolescents (12 to 17 years old), of which 15 received placebo and 15 received depemokimab 100 mg subcutaneously. In a combined analysis of these studies, a 43% reduction in clinically significant exacerbations was observed in adolescents following depemokimab treatment compared to placebo (rate ratio 0.57; 95% CI: 0.15, 2.13).
Chronic rhinosinusitis with nasal polyps (CRSwNP)
There are no clinical data available in children and adolescents aged less than 18 years old.
The European Medicines Agency has waived the obligation to submit the results of studies with EXDENSUR in all subsets of the paediatric population in asthma and CRSwNP (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Depemokimab exhibited approximately dose-proportional pharmacokinetics over a dose range of 10 to 300 mg in patients with asthma following subcutaneous administration. After subcutaneous administration of 100 mg depemokimab every 6 months, the average concentrations (%CV) at steady state were 5.9 mcg/mL (28%) and 5.2 mcg/mL (27%) in asthma and CRSwNP patients, respectively. The Week 26 trough concentrations were 1.2 mcg/mL (37%) and 1.0 mcg/mL (36%) in asthma and CRSwNP patients, respectively.
Absorption
Following a single subcutaneous administration (doses ranging from 2 to 300 mg), maximum observed plasma concentrations (Cmax) were achieved at a median time ranging from 8 to 14 days. After a single subcutaneous administration of 100 mg depemokimab, the average Cmax (%CV) was 12.2 mcg/mL (16%).
Following two repeat subcutaneous administrations once every 6 months, achieving steady-state conditions, the depemokimab accumulation was negligible (<10%).
Distribution
Following a single subcutaneous administration of depemokimab, the mean apparent volume of distribution is 6 to 9 L.
Biotransformation
Depemokimab is a monoclonal antibody which is catabolised by ubiquitous proteolytic enzymes not restricted to hepatic tissue.
Elimination
Following a single subcutaneous administration of depemokimab, the geometric mean terminal half-life ranged from 38 to 53 days, with geometric mean apparent clearance values ranging from 0.081 to 0.16 L/day.
Special populations
Body weight
Population pharmacokinetic analyses indicated that exposure to depemokimab decreases with increasing body weight. Weight was the major determinant of depemokimab exposure, and satisfied conventional allometry with coefficients of 0.841 for CL/F and 0.887 for V/F, typical for a mAb such as depemokimab. Over the body weight range of 54 to 108 kg (corresponding to the 5th to 95th percentiles), the difference in all exposure metrics was less than 1.3-fold. Thus, the magnitude of effect of body weight on depemokimab exposure is not considered clinically relevant within this body weight range. At high body weights (140-160 kg) the exposure may be decreased 2-fold. For such patients, reduced efficacy cannot be excluded.
Gender, ethnicity
Population pharmacokinetic analyses indicated there was no clinically relevant effect of gender or race on depemokimab pharmacokinetics.
Elderly
Available pharmacokinetic data in elderly patients (≥65 years old, N=176) across all clinical studies showed that depemokimab pharmacokinetics were similar between adult patients and patients aged 65 years and older (up to 93 years), based on the population pharmacokinetic analysis.
Renal impairment
No formal studies have been conducted to investigate the effect of renal impairment on the pharmacokinetics of depemokimab. Based on population pharmacokinetic analyses, no dose adjustment is required in patients with impaired renal function. Data are limited (n=2) in patients with an eGFR <30 mL/min/1.73m², but CL/F values of these two patients fell within the range of patients with normal renal function.
Renal impairment is not expected to have a significant impact on clearance as depemokimab is not cleared renally.
Hepatic impairment
No formal studies have been conducted to investigate the effect of hepatic impairment on the pharmacokinetics of depemokimab. Since depemokimab is degraded by widely distributed proteolytic enzymes, not restricted to hepatic tissue, changes in hepatic function are unlikely to have any effect on the elimination of depemokimab. Based on population pharmacokinetic analysis, baseline hepatic function biomarkers (alanine aminotransferase [ALT], aspartate aminotransferase [AST] and bilirubin) had no clinically relevant effect on depemokimab apparent clearance.
Paediatric population
Asthma:
There are limited pharmacokinetic data available in the paediatric population (15 adolescent patients with asthma). The pharmacokinetics of depemokimab in adolescents aged 12 to 17 years were similar to adults (see section 4.2) and key pharmacokinetics parameters are presented in Table 7.
Table 7. Derived secondary pharmacokinetics parameters in patients who received depemokimab 100 mg subcutaneously every 26 weeks in the pooled SWIFT-1 and SWIFT-2 studies:
| Parameter - geometric mean (%CV) | Adolescents N=15 | Adults N=479 | Overall N=494 |
| AUCtau,ss (mcg*day/mL) | 1051 (31) | 1082 (28) | 1081 (28) |
| Cav,ss (mcg /mL) | 5.8 (31) | 5.9 (28) | 5.9 (28) |
| Cmax,26-52 (mcg/mL) | 14.6 (30) | 13.6 (28) | 13.6 (28) |
| Tmax,26-52 (day) | 10.8 (9) | 13.7 (18) | 13.6 (18) |
| Ctrough,week52 (mcg/mL) | 1.1 (39) | 1.3 (38) | 1.3 (38) |
| t1/2 (days) | 44.7 (9) | 48.7 (10) | 48.6 (10) |
AUCtau,ss; area under the concentration-time curve during a dosing interval at steady state, Cav,ss; average concentration during a dosing interval, Cmax,26-52; maximum concentration during the second dosing interval, Tmax,26-52; time to maximum concentration during the second dosing interval, Ctrough,week52; trough concentration at the end of the second administration, t1/2; half-life
The pharmacokinetics of depemokimab have not been established in paediatric patients with asthma aged less than 12 years of age.
Pharmacokinetic/pharmacodynamic relationship
There is a clear relationship between depemokimab pharmacokinetics and reduction in blood eosinophil counts (pharmacodynamics) with maximum achievable reduction of around 85% and a half-maximal effective concentration (EC50) of 0.19 mcg/mL. The concentration associated with 90% of maximal effect (EC90) was 0.75 mcg/mL.
5.3. Preclinical safety data
Non-clinical data reveal no special hazard for humans based on studies of repeated dose toxicity with safety pharmacology endpoints.
No genotoxicity, carcinogenicity or reproductive toxicology studies have been conducted with depemokimab.
In animal studies targeting IL-5 signaling pathways (e.g. knockout animal data and class effects), there were no developmental effects observed.
Male and female fertility are unlikely to be affected based upon no adverse histopathological findings in the reproductive organs from cynomolgus monkeys at exposures sufficiently in excess of the maximum human exposure. Mating and reproductive performance were unaffected in male and female CD-1 mice receiving an analogous antibody, which inhibits the activity of murine IL-5.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.