EXTAVIA Powder and solvent for solution for injection Ref.[8863] Active ingredients: Interferon beta-1b
Source: European Medicines Agency (EU) Revision Year: 2019 Publisher: Novartis Europharm Limited, Vista Building, Elm Park, Merrion Road, Dublin 4, Ireland
Contraindications
- Hypersensitivity to natural or recombinant interferon beta, human albumin or to any of the excipients listed in section 6.1.
- Patients with current severe depression and/or suicidal ideation (see sections 4.4 and 4.8).
- Patients with decompensated liver disease (see sections 4.4, 4.5 and 4.8).
Special warnings and precautions for use
Traceability
In order to improve traceability of biological medicinal products, the name and the batch number of the administered product should be clearly recorded.
Immune system disorders
The administration of cytokines to patients with a pre-existing monoclonal gammopathy has been associated with the development of systemic capillary leak syndrome with shock-like symptoms and fatal outcome.
Gastrointestinal disorders
Cases of pancreatitis were observed with Extavia use, often associated with hypertriglyceridaemia.
Nervous system disorders
Extavia should be administered with caution to patients with previous or current depressive disorders, in particular to those with antecedents of suicidal ideation (see section 4.3). Depression and suicidal ideation are known to occur with increased frequency in the multiple sclerosis population and in association with interferon use. Patients treated with Extavia should be advised to immediately report any symptoms of depression and/or suicidal ideation to their prescribing physician. Patients exhibiting depression should be monitored closely during therapy with Extavia and treated appropriately. Cessation of therapy with Extavia should be considered (see also sections 4.3 and 4.8).
Extavia should be administered with caution to patients with a history of seizures, to patients receiving treatment with anti-epileptics, and in particular to patients with epilepsy who are not adequately controlled with anti-epileptics (see sections 4.5 and 4.8).
This medicinal product contains human albumin and hence carries a potential risk for transmission of viral diseases. A risk for transmission of Creutzfeld-Jacob disease (CJD) cannot be excluded.
Laboratory tests
Regular thyroid function tests are recommended in patients with a history of thyroid dysfunction or as clinically indicated.
In addition to those laboratory tests normally required for monitoring patients with multiple sclerosis, complete blood and differential white blood cell counts, platelet counts, and blood chemistries, including liver function tests (e.g. aspartate aminotransferase serum glutamic-oxaloacetic transaminase (SGOT), alanine aminotransferase serum glutamate pyruvate transaminase (SGPT) and gamma glutamyltransferase), are recommended prior to initiation and at regular intervals following introduction of Extavia therapy, and then periodically thereafter in the absence of clinical symptoms.
Patients with anaemia, thrombocytopenia or leukopenia (alone or in any combination) may require more intensive monitoring of complete blood cell counts, with differential and platelet counts. Patients who develop neutropenia should be monitored closely for the development of fever or infection. There have been reports of thrombocytopenia, with profound decreases in platelet count.
Hepatobiliary disorders
Asymptomatic elevations of serum transaminases, in most cases mild and transient, occurred very commonly in patients treated with Extavia during clinical trials. As for other beta interferons, cases of severe hepatic injury, including hepatic failure, have been reported in patients treated with Extavia. The most serious events often occurred in patients exposed to other medicinal products or substances known to be associated with hepatotoxicity or in the presence of co-morbid medical conditions (e.g. metastasising malignant disease, severe infection and sepsis, alcohol abuse).
Patients should be monitored for signs of hepatic injury. The occurrence of elevations in serum transaminases should lead to close monitoring and investigation. Withdrawal of Extavia should be considered if the levels significantly increase or if they are associated with clinical symptoms such as jaundice. In the absence of clinical evidence for liver damage, and after normalisation of liver enzymes, a reintroduction of therapy could be considered with appropriate follow-up of hepatic functions.
Thrombotic microangiopathy (TMA)
Cases of thrombotic microangiopathy, manifested as thrombotic thrombocytopenic purpura (TTP) or haemolytic uraemic syndrome (HUS), including fatal cases, have been reported with interferon beta products. Events were reported at various time points during treatment and may occur several weeks to several years after starting treatment with interferon beta. Early clinical features include thrombocytopenia, new onset hypertension, fever, central nervous system symptoms (e.g. confusion, paresis) and impaired renal function. Laboratory findings suggestive of TMA include decreased platelet counts, increased serum lactate dehydrogenase (LDH) due to haemolysis and schistocytes (erythrocyte fragmentation) on a blood film. Therefore if clinical features of TMA are observed, further testing of blood platelet levels, serum LDH, blood films and renal function is recommended. If TMA is diagnosed, prompt treatment is required (considering plasma exchange) and immediate discontinuation of Extavia is recommended.
Renal and urinary disorders
Caution should be used and close monitoring considered when administering interferon beta to patients with severe renal failure.
Nephrotic syndrome
Cases of nephrotic syndrome with different underlying nephropathies including collapsing focal segmental glomerulosclerosis (FSGS), minimal change disease (MCD), membranoproliferative glomerulonephritis (MPGN) and membranous glomerulopathy (MGN) have been reported during treatment with interferon beta products. Events were reported at various time points during treatment and may occur after several years of treatment with interferon beta. Periodic monitoring of early signs or symptoms, e.g. oedema, proteinuria and impaired renal function, is recommended, especially in patients at higher risk of renal disease. Prompt treatment of nephrotic syndrome is required and discontinuation of treatment with Extavia should be considered.
Cardiac disorders
Extavia should also be used with caution in patients who suffer from pre-existing cardiac disorders. Patients with pre-existing significant cardiac disease, such as congestive heart failure, coronary artery disease or arrhythmia, should be monitored for worsening of their cardiac condition, particularly during initiation of treatment with Extavia.
While Extavia does not have any known direct-acting cardiac toxicity, symptoms of the flu-like syndrome associated with beta interferons may prove stressful to patients with pre-existing significant cardiac disease. During the post-marketing period very rare reports have been received of temporary worsening of cardiac status at the start of Extavia therapy in patients with pre-existing significant cardiac disease.
Cases of cardiomyopathy have been reported. If this occurs and a relationship to Extavia is suspected, treatment should be discontinued.
General disorders and administration site conditions
Serious hypersensitivity reactions (severe acute reactions such as bronchospasm, anaphylaxis and urticaria) may occur. If reactions are severe, Extavia should be discontinued and appropriate medical intervention instituted.
Injection site necrosis has been reported in patients using Extavia (see section 4.8). It can be extensive and may involve muscle fascia as well as fat and therefore can result in scar formation. Debridement and, less often, skin grafting are occasionally required and healing may take up to 6 months.
If the patient experiences any break in the skin, which may be associated with swelling or drainage of fluid from the injection site, the patient should be advised to consult with his/her physician before continuing injections with Extavia.
If the patient has multiple lesions Extavia should be discontinued until healing has occurred. Patients with single lesions may continue on Extavia provided the necrosis is not too extensive, as some patients have experienced healing of injection site necrosis whilst on Extavia.
To minimise the risk of injection site necrosis patients should be advised to:
- use an aseptic injection technique,
- rotate the injection sites with each dose.
The incidence of injection site reactions may be reduced by the use of an auto-injector. In the pivotal study of patients with a single clinical event suggestive of multiple sclerosis an auto-injector was used in the majority of patients. Injection site reactions and necroses were observed less frequently in this study than in the other pivotal studies.
The procedure for self-administration by the patient should be reviewed periodically, especially if injection site reactions have occurred.
Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity. In controlled clinical trials serum samples were collected every 3 months for monitoring of development of antibodies to Extavia.
In the different controlled clinical trials, between 23% and 41% of the patients developed serum interferon beta-1b neutralising activity confirmed by at least two consecutive positive titres. Between 43% and 55% of these patients converted to a stable antibody negative status (based on two consecutive negative titres) during the subsequent observational period of the trial concerned.
The development of neutralising activity is associated with a reduction in clinical efficacy only with regard to relapse activity. Some analyses suggest that this effect might be more pronounced in patients with higher titre levels of neutralising activity.
In the study of patients with a single clinical event suggestive of multiple sclerosis, neutralising activity measured every 6 months was observed at least once in 32% (89) of the patients treated immediately with Extavia. 60% (53) of these patients returned to negative status based on the last available assessment within the 5-year period. Within this period, the development of neutralising activity was associated with a significant increase in newly active lesions and T2 lesion volume on magnetic resonance imaging. However, this did not seem to be associated with a reduction in clinical efficacy (with regard to time to clinically definite multiple sclerosis (CDMS), time to confirmed EDSS progression and relapse rate).
New adverse events have not been associated with the development of neutralising activity.
It has been demonstrated in vitro that Extavia cross-reacts with natural interferon beta. However, this has not been investigated in vivo and its clinical significance is uncertain.
There are sparse and inconclusive data on patients who have developed neutralising activity and have completed Extavia therapy.
The decision to continue or discontinue treatment should be based on clinical disease activity rather than on neutralising activity status.
Excipients
This medicinal product contains less than 1 mmol sodium (23 mg) per ml, i.e. essentially 'sodiumfree'.
Latex-sensitive individuals
The removable tip cap of the Extavia pre-filled syringe contains a derivative of natural rubber latex. Although no natural rubber latex is detected in the cap, the safe use of Extavia pre-filled syringe in latex-sensitive individuals has not been studied and there is therefore a potential risk for hypersensitivity reactions which cannot be completely ruled out.
Interaction with other medicinal products and other forms of interaction
No interaction studies have been performed.
The effect of alternate-day administration of 250 microgram (8.0 million IU) Extavia on drug metabolism in multiple sclerosis patients is unknown. Corticosteroid or ACTH treatment of relapses for periods of up to 28 days has been well tolerated in patients receiving Extavia.
Due to the lack of clinical experience in multiple sclerosis patients, the use of Extavia together with immunomodulators other than corticosteroids or ACTH is not recommended.
Interferons have been reported to reduce the activity of hepatic cytochrome P450-dependent enzymes in humans and animals. Caution should be exercised when Extavia is administered in combination with medicinal products that have a narrow therapeutic index and are largely dependent on the hepatic cytochrome P450 system for clearance, e.g. anti-epileptics. Additional caution should be exercised with any co-medication which has an effect on the haematopoetic system.
Fertility, pregnancy and lactation
Pregnancy
A large amount of data (more than 1,000 pregnancy outcomes) from interferon beta registries, national registries and post-marketing experience indicates no increased risk of major congenital anomalies, after pre-conception exposure or exposure during the first trimester of pregnancy.
However, the duration of exposure during the first trimester is uncertain, because data were collected when interferon beta use was contraindicated during pregnancy, and treatment was likely interrupted when the pregnancy was detected and/or confirmed. Experience with exposure during the second and third trimesters is very limited.
Based on animal data (see section 5.3), there is a possibly increased risk for spontaneous abortion. The risk of spontaneous abortions in pregnant women exposed to interferon beta cannot adequately be evaluated by means of the currently available data, but the data suggest no increased risk so far. If clinically needed, the use of Extavia may be considered during pregnancy.
Breast-feeding
Limited information available on the transfer of interferon beta-1b into breast milk, together with the chemical/physiological characteristics of interferon beta, suggests that levels of interferon beta-1b excreted in human milk are negligible. No harmful effects on the breast-fed newborn/infant are anticipated.
Extavia can be used during breast-feeding.
Fertility
No investigations on fertility have been conducted (see section 5.3).
Effects on ability to drive and use machines
No studies on the effects on the ability to drive and use machines have been performed.
Adverse events related to the central nervous system associated with the use of Extavia might influence the ability to drive and use machines in susceptible patients.
Undesirable effects
Summary of the safety profile
At the beginning of treatment adverse reactions are common but in general they subside with further treatment. The most frequently observed adverse reactions are a flu-like symptom complex (fever, chills, arthralgia, malaise, sweating, headache, or myalgia), which is mainly due to the pharmacological effects of the medicinal product, and injection site reactions. Injection site reactions occurred frequently after administration of Extavia. Redness, swelling, discolouration, inflammation, pain, hypersensitivity, necrosis and non-specific reactions were significantly associated with 250 microgram (8.0 million IU) Extavia treatment.
Generally, dose titration is recommended at the start of treatment in order to increase tolerability to Extavia (see section 4.2). Flu-like symptoms may also be reduced by administration of non-steroidal anti-inflammatory medicinal products. The incidence of injection site reactions may be reduced by the use of an auto-injector.
Tabulated list of adverse reactions
In the following tables, the most appropriate MedDRA term is used to describe a certain reaction and its synonyms and related conditions.
The following adverse event listings are based on reports from clinical trials (Table 1, adverse events and laboratory abnormalities) and from post-marketing surveillance (Table 2, frequencies - where known - based on pooled clinical trials (very common ≥1/10, common ≥1/100 to <1/10, uncommon ≥1/1,000 to <1/100, rare ≥1/10,000 to <1/1,000, very rare <1/10,000)) of Extavia use. Experience with Extavia in patients with multiple sclerosis (MS) is limited, consequently those adverse events which occur very rarely may not yet have been observed.
Table 1. Adverse events and laboratory abnormalities with incidence rates ≥10% and the respective percentages under placebo; significantly associated side effects <10% based on reports from clinical trials:
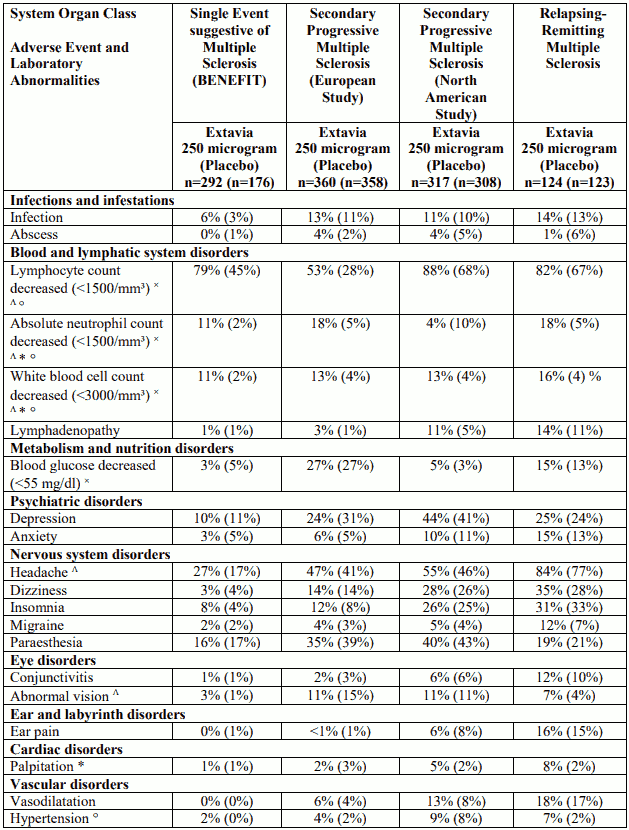
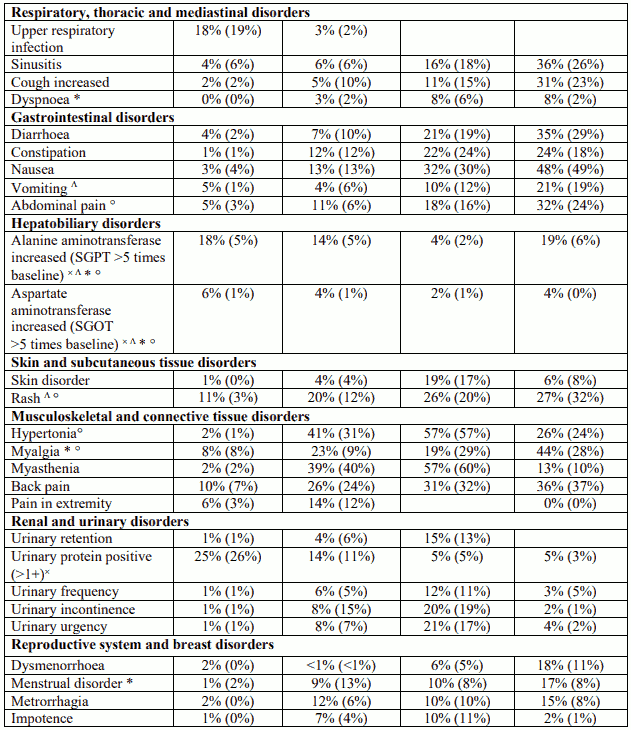
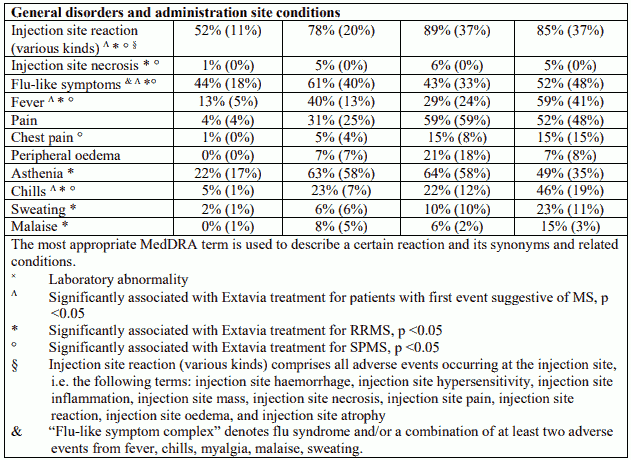
Table 2. Adverse drug reactions (ADRs) identified during post-marketing surveillance (frequencies - where known - calculated based on pooled clinical trial data N=1,093):
Blood and lymphatic system disorders
Common: Anaemia
Uncommon: Thrombocytopenia
Rare: Thrombotic microangiopathy including thrombotic thrombocytopenic purpura/haemolytic uraemic syndrome#
Immune system disorders
Rare: Anaphylactic reactions
Νot known: Capillary leak syndrome in preexisting monoclonal gammopathy*
Endocrine disorders
Common: Hypothyroidism
Rare: Hyperthyroidism, Thyroid disorders
Metabolism and nutrition disorders
Common: Weight increased, Weight decreased
Uncommon: Blood triglycerides increased
Rare: Anorexia*
Psychiatric disorders
Common: Confusional state
Uncommon: Suicide attempt (see also section 4.4), Emotional lability
Nervous system disorders
Uncommon: Convulsion
Cardiac disorders
Common: Tachycardia
Rare: Cardiomyopathy*
Respiratory, thoracic and mediastinal disorders
Rare: Bronchospasm*
Νot known: Pulmonary arterial hypertension**
Gastrointestinal disorders
Rare: Pancreatitis
Hepatobiliary disorders
Common: Blood bilirubin increased
Uncommon: Gamma-glutamyltransferase increased, Hepatitis
Rare: Hepatic injury (including hepatitis), Hepatic failure*
Skin and subcutaneous tissue disorders
Common: Urticaria, Pruritus, Alopecia
Uncommon: Skin discolouration
Musculoskeletal and connective tissue disorders
Very common: Arthralgia
Νot known: Drug-induced lupus erythematosus
Renal and urinary disorders
Uncommon: Nephrotic syndrome, glomerulosclerosis (see section 4.4)*,#
Reproductive system and breast disorders
Common: Menorrhagia
* ADRs derived only during post-marketing.
# Class label for interferon beta products (see section 4.4).
** Class label for interferon products, see below "Pulmonary arterial hypertension".
Pulmonary arterial hypertension
Cases of pulmonary arterial hypertension (PAH) have been reported with interferon beta products. Events were reported at various time points including up to several years after starting treatment with interferon beta.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed in Appendix V.
Incompatibilities
This medicinal product must not be mixed with other medicinal products except for the supplied solvent mentioned in section 6.6.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.