FLOMAX Capsule Ref.[10563] Active ingredients: Tamsulosin
Source: FDA, National Drug Code (US) Revision Year: 2020
12.1. Mechanism of Action
The symptoms associated with benign prostatic hyperplasia (BPH) are related to bladder outlet obstruction, which is comprised of two underlying components: static and dynamic. The static component is related to an increase in prostate size caused, in part, by a proliferation of smooth muscle cells in the prostatic stroma. However, the severity of BPH symptoms and the degree of urethral obstruction do not correlate well with the size of the prostate. The dynamic component is a function of an increase in smooth muscle tone in the prostate and bladder neck leading to constriction of the bladder outlet. Smooth muscle tone is mediated by the sympathetic nervous stimulation of alpha1 adrenoceptors, which are abundant in the prostate, prostatic capsule, prostatic urethra, and bladder neck. Blockade of these adrenoceptors can cause smooth muscles in the bladder neck and prostate to relax, resulting in an improvement in urine flow rate and a reduction in symptoms of BPH.
Tamsulosin, an alpha1 adrenoceptor blocking agent, exhibits selectivity for alpha1 receptors in the human prostate. At least three discrete alpha1 adrenoceptor subtypes have been identified: alpha1A, alpha1B, and alpha1D; their distribution differs between human organs and tissue. Approximately 70% of the alpha1 receptors in the human prostate are of the alpha1A subtype.
FLOMAX capsules are not intended for use as an antihypertensive drug.
12.2. Pharmacodynamics
Urologic pharmacodynamic effects have been evaluated in neurologically impaired pediatric patients and in adults with BPH [see Use in Specific Populations (8.4) and Clinical Studies (14)].
12.3. Pharmacokinetics
The pharmacokinetics of tamsulosin hydrochloride have been evaluated in adult healthy volunteers and patients with BPH after single and/or multiple administration with doses ranging from 0.1 mg to 1 mg.
Absorption
Absorption of tamsulosin hydrochloride from FLOMAX capsules 0.4 mg is essentially complete (>90%) following oral administration under fasting conditions. Tamsulosin hydrochloride exhibits linear kinetics following single and multiple dosing, with achievement of steady-state concentrations by the fifth day of once-a-day dosing.
Effect of Food
The time to maximum concentration (Tmax) is reached by 4 to 5 hours under fasting conditions and by 6 to 7 hours when FLOMAX capsules are administered with food. Taking FLOMAX capsules under fasted conditions results in a 30% increase in bioavailability (AUC) and 40% to 70% increase in peak concentrations (Cmax) compared to fed conditions (Figure 1).
Figure 1. Mean Plasma Tamsulosin Hydrochloride Concentrations Following Single-Dose Administration of FLOMAX Capsules 0.4 mg Under Fasted and Fed Conditions (n=8):
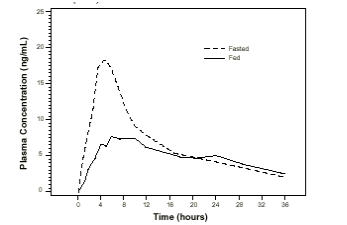
The effects of food on the pharmacokinetics of tamsulosin hydrochloride are consistent regardless of whether a FLOMAX capsule is taken with a light breakfast or a high-fat breakfast (Table 2).
Table 2 Mean (± S.D.) Pharmacokinetic Parameters Following FLOMAX Capsules 0.4 mg Once Daily or 0.8 mg Once Daily with a Light Breakfast, High-Fat Breakfast or Fasted:
| Pharmacokinetic Parameter | 0.4 mg QD to healthy volunteers; n=23 (age range 18-32 years) | 0.8 mg QD to healthy volunteers; n=22 (age range 55-75 years) | |||
|---|---|---|---|---|---|
| Light Breakfast | Fasted | Light Breakfast | High-Fat Breakfast | Fasted | |
| Cmin (ng/mL) | 4.0 ± 2.6 | 3.8 ± 2.5 | 12.3 ± 6.7 | 13.5 ± 7.6 | 13.3 ± 13.3 |
| Cmax (ng/mL) | 10.1 ± 4.8 | 17.1 ± 17.1 | 29.8 ± 10.3 | 29.1 ± 11.0 | 41.6 ± 15.6 |
| Cmax/Cmin Ratio | 3.1 ± 1.0 | 5.3 ± 2.2 | 2.7 ± 0.7 | 2.5 ± 0.8 | 3.6 ± 1.1 |
| Tmax (hours) | 6.0 | 4.0 | 7.0 | 6.6 | 5.0 |
| T1/2 (hours) | - | - | - | - | 14.9 ± 3.9 |
| AUCτ (ng•hr/mL) | 151 ± 81.5 | 199 ± 94.1 | 440 ± 195 | 449 ± 217 | 557 ± 257 |
Cmin = observed minimum concentration
Cmax = observed maximum tamsulosin hydrochloride plasma concentration
Tmax = median time-to-maximum concentration
T1/2 = observed half-life
AUCτ = area under the tamsulosin hydrochloride plasma time curve over the dosing interval
Distribution
The mean steady-state apparent volume of distribution of tamsulosin hydrochloride after intravenous administration to 10 healthy male adults was 16 L, which is suggestive of distribution into extracellular fluids in the body.
Tamsulosin hydrochloride is extensively bound to human plasma proteins (94% to 99%), primarily alpha1 acid glycoprotein (AAG), with linear binding over a wide concentration range (20 to 600 ng/mL). The results of two-way in vitro studies indicate that the binding of tamsulosin hydrochloride to human plasma proteins is not affected by amitriptyline, diclofenac, glyburide, simvastatin plus simvastatin-hydroxy acid metabolite, warfarin, diazepam, propranolol, trichlormethiazide, or chlormadinone. Likewise, tamsulosin hydrochloride had no effect on the extent of binding of these drugs.
Metabolism
There is no enantiomeric bioconversion from tamsulosin hydrochloride [R(-) isomer] to the S(+) isomer in humans. Tamsulosin hydrochloride is extensively metabolized by cytochrome P450 enzymes in the liver and less than 10% of the dose is excreted in urine unchanged. However, the pharmacokinetic profile of the metabolites in humans has not been established. Tamsulosin is extensively metabolized, mainly by CYP3A4 and CYP2D6 as well as via some minor participation of other CYP isoenzymes. Inhibition of hepatic drug-metabolizing enzymes may lead to increased exposure to tamsulosin [see Warnings and Precautions (5.2) and Drug Interactions (7.1)]. The metabolites of tamsulosin hydrochloride undergo extensive conjugation to glucuronide or sulfate prior to renal excretion.
Incubations with human liver microsomes showed no evidence of clinically significant metabolic interactions between tamsulosin hydrochloride and amitriptyline, albuterol (beta agonist), glyburide (glibenclamide) and finasteride (5alpha-reductase inhibitor for treatment of BPH). However, results of the in vitro testing of the tamsulosin hydrochloride interaction with diclofenac and warfarin were equivocal.
Excretion
On administration of the radiolabeled dose of tamsulosin hydrochloride to 4 healthy volunteers, 97% of the administered radioactivity was recovered, with urine (76%) representing the primary route of excretion compared to feces (21%) over 168 hours.
Following intravenous or oral administration of an immediate-release formulation, the elimination half-life of tamsulosin hydrochloride in plasma ranged from 5 to 7 hours. Because of absorption rate-controlled pharmacokinetics with FLOMAX capsules, the apparent half-life of tamsulosin hydrochloride is approximately 9 to 13 hours in healthy volunteers and 14 to 15 hours in the target population.
Tamsulosin hydrochloride undergoes restrictive clearance in humans, with a relatively low systemic clearance (2.88 L/h).
Specific Populations
Pediatric Use
FLOMAX capsules are not indicated for use in pediatric populations [see Use in Specific Populations (8.4)].
Geriatric (Age) Use
Cross-study comparison of FLOMAX capsules overall exposure (AUC) and half-life indicates that the pharmacokinetic disposition of tamsulosin hydrochloride may be slightly prolonged in geriatric males compared to young, healthy male volunteers. Intrinsic clearance is independent of tamsulosin hydrochloride binding to AAG, but diminishes with age, resulting in a 40% overall higher exposure (AUC) in subjects of age 55 to 75 years compared to subjects of age 20 to 32 years [see Use in Specific Populations (8.5)].
Renal Impairment
The pharmacokinetics of tamsulosin hydrochloride have been compared in 6 subjects with mild-moderate (30≤ CLcr <70 mL/min/1.73 m²) or moderate-severe (10≤ CLcr <30 mL/min/1.73 m²) renal impairment and 6 normal subjects (CLcr >90 mL/min/1.73 m²). While a change in the overall plasma concentration of tamsulosin hydrochloride was observed as the result of altered binding to AAG, the unbound (active) concentration of tamsulosin hydrochloride, as well as the intrinsic clearance, remained relatively constant. Therefore, patients with renal impairment do not require an adjustment in FLOMAX capsules dosing. However, patients with end-stage renal disease (CLcr <10 mL/min/1.73 m²) have not been studied [see Use in Specific Populations (8.6)].
Hepatic Impairment
The pharmacokinetics of tamsulosin hydrochloride have been compared in 8 subjects with moderate hepatic impairment (Child-Pugh's classification: Grades A and B) and 8 normal subjects. While a change in the overall plasma concentration of tamsulosin hydrochloride was observed as the result of altered binding to AAG, the unbound (active) concentration of tamsulosin hydrochloride does not change significantly, with only a modest (32%) change in intrinsic clearance of unbound tamsulosin hydrochloride. Therefore, patients with moderate hepatic impairment do not require an adjustment in FLOMAX capsules dosage. FLOMAX has not been studied in patients with severe hepatic impairment [see Use in Specific Populations (8.7)].
Drug Interactions
Cytochrome P450 Inhibition
Strong and Moderate Inhibitors of CYP3A4 or CYP2D6:
The effects of ketoconazole (a strong inhibitor of CYP3A4) at 400 mg once daily for 5 days on the pharmacokinetics of a single FLOMAX capsule 0.4 mg dose was investigated in 24 healthy volunteers (age range 23 to 47 years). Concomitant treatment with ketoconazole resulted in an increase in the Cmax and AUC of tamsulosin by a factor of 2.2 and 2.8, respectively [see Warnings and Precautions (5.2) and Clinical Pharmacology (12.3)]. The effects of concomitant administration of a moderate CYP3A4 inhibitor (e.g., erythromycin) on the pharmacokinetics of FLOMAX have not been evaluated [see Warnings and Precautions (5.2) and Drug Interactions (7.1)].
The effects of paroxetine (a strong inhibitor of CYP2D6) at 20 mg once daily for 9 days on the pharmacokinetics of a single FLOMAX capsule 0.4 mg dose was investigated in 24 healthy volunteers (age range 23 to 47 years). Concomitant treatment with paroxetine resulted in an increase in the Cmax and AUC of tamsulosin by a factor of 1.3 and 1.6, respectively [see Warnings and Precautions (5.2) and Drug Interactions (7.1)]. A similar increase in exposure is expected in CYP2D6 poor metabolizers (PM) as compared to extensive metabolizers (EM). A fraction of the population (about 7% of Caucasians and 2% of African Americans) are CYP2D6 PMs. Since CYP2D6 PMs cannot be readily identified and the potential for significant increase in tamsulosin exposure exists when FLOMAX 0.4 mg is co-administered with strong CYP3A4 inhibitors in CYP2D6 PMs, FLOMAX 0.4 mg capsules should not be used in combination with strong inhibitors of CYP3A4 (e.g., ketoconazole) [see Warnings and Precautions (5.2) and Drug Interactions (7.1)].
The effects of concomitant administration of a moderate CYP2D6 inhibitor (e.g., terbinafine) on the pharmacokinetics of FLOMAX have not been evaluated [see Warnings and Precautions (5.2) and Drug Interactions (7.1)].
The effects of co-administration of both a CYP3A4 and a CYP2D6 inhibitor with FLOMAX capsules have not been evaluated. However, there is a potential for significant increase in tamsulosin exposure when FLOMAX 0.4 mg is co-administered with a combination of both CYP3A4 and CYP2D6 inhibitors [see Warnings and Precautions (5.2) and Drug Interactions (7.1)].
Cimetidine
The effects of cimetidine at the highest recommended dose (400 mg every 6 hours for 6 days) on the pharmacokinetics of a single FLOMAX capsule 0.4 mg dose was investigated in 10 healthy volunteers (age range 21 to 38 years). Treatment with cimetidine resulted in a significant decrease (26%) in the clearance of tamsulosin hydrochloride, which resulted in a moderate increase in tamsulosin hydrochloride AUC (44%) [see Warnings and Precautions (5.2) and Drug Interactions (7.1)].
Other Alpha Adrenergic Blocking Agents
The pharmacokinetic and pharmacodynamic interactions between FLOMAX capsules and other alpha adrenergic blocking agents have not been determined; however, interactions between FLOMAX capsules and other alpha adrenergic blocking agents may be expected [see Warnings and Precautions (5.2) and Drug Interactions (7.2)].
PDE5 Inhibitors
Caution is advised when alpha adrenergic blocking agents, including FLOMAX, are co-administered with PDE5 inhibitors. Alpha-adrenergic blockers and PDE5 inhibitors are both vasodilators that can lower blood pressure. Concomitant use of these two drug classes can potentially cause symptomatic hypotension [see Warnings and Precautions (5.2) and Drug Interactions (7.3)].
Warfarin
A definitive drug-drug interaction study between tamsulosin hydrochloride and warfarin was not conducted. Results from limited in vitro and in vivo studies are inconclusive. Therefore, caution should be exercised with concomitant administration of warfarin and FLOMAX capsules [see Warnings and Precautions (5.2) and Drug Interactions (7.4)].
Nifedipine, Atenolol, Enalapril
In three studies in hypertensive subjects (age range 47 to 79 years) whose blood pressure was controlled with stable doses of nifedipine, atenolol, or enalapril for at least 3 months, FLOMAX capsules 0.4 mg for 7 days followed by FLOMAX capsules 0.8 mg for another 7 days (n=8 per study) resulted in no clinically significant effects on blood pressure and pulse rate compared to placebo (n=4 per study). Therefore, dosage adjustments are not necessary when FLOMAX capsules are administered concomitantly with nifedipine, atenolol, or enalapril [see Drug Interactions (7.5)].
Digoxin and Theophylline
In two studies in healthy volunteers (n=10 per study; age range 19 to 39 years) receiving FLOMAX capsules 0.4 mg/day for 2 days, followed by FLOMAX capsules 0.8 mg/day for 5 to 8 days, single intravenous doses of digoxin 0.5 mg or theophylline 5 mg/kg resulted in no change in the pharmacokinetics of digoxin or theophylline. Therefore, dosage adjustments are not necessary when a FLOMAX capsule is administered concomitantly with digoxin or theophylline [see Drug Interactions (7.6)].
Furosemide
The pharmacokinetic and pharmacodynamic interaction between FLOMAX capsules 0.8 mg/day (steady-state) and furosemide 20 mg intravenously (single dose) was evaluated in 10 healthy volunteers (age range 21 to 40 years). FLOMAX capsules had no effect on the pharmacodynamics (excretion of electrolytes) of furosemide. While furosemide produced an 11% to 12% reduction in tamsulosin hydrochloride Cmax and AUC, these changes are expected to be clinically insignificant and do not require adjustment of the FLOMAX capsules dosage [see Drug Interactions (7.7)].
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Rats administered doses up to 43 mg/kg/day in males and 52 mg/kg/day in females had no increases in tumor incidence, with the exception of a modest increase in the frequency of mammary gland fibroadenomas in female rats receiving doses ≥5.4 mg/kg (P<0.015). The highest doses of tamsulosin hydrochloride evaluated in the rat carcinogenicity study produced systemic exposures (AUC) in rats 3 times the exposures in men receiving the maximum therapeutic dose of 0.8 mg/day.
Mice were administered doses up to 127 mg/kg/day in males and 158 mg/kg/day in females. There were no significant tumor findings in male mice. Female mice treated for 2 years with the two highest doses of 45 and 158 mg/kg/day had statistically significant increases in the incidence of mammary gland fibroadenomas (P<0.0001) and adenocarcinomas (P<0.0075). The highest dose levels of tamsulosin hydrochloride evaluated in the mice carcinogenicity study produced systemic exposures (AUC) in mice 8 times the exposures in men receiving the maximum therapeutic dose of 0.8 mg/day.
The increased incidences of mammary gland neoplasms in female rats and mice were considered secondary to tamsulosin hydrochloride-induced hyperprolactinemia. It is not known if FLOMAX capsules elevate prolactin in humans. The relevance for human risk of the findings of prolactin-mediated endocrine tumors in rodents is not known.
Tamsulosin hydrochloride produced no evidence of mutagenic potential in vitro in the Ames reverse mutation test, mouse lymphoma thymidine kinase assay, unscheduled DNA repair synthesis assay, and chromosomal aberration assays in Chinese hamster ovary cells or human lymphocytes. There were no mutagenic effects in the in vivo sister chromatid exchange and mouse micronucleus assay.
Studies in rats revealed significantly reduced fertility in males dosed with single or multiple daily doses of 300 mg/kg/day of tamsulosin hydrochloride (AUC exposure in rats about 50 times the human exposure with the maximum therapeutic dose). The mechanism of decreased fertility in male rats is considered to be an effect of the compound on the vaginal plug formation possibly due to changes of semen content or impairment of ejaculation. The effects on fertility were reversible, showing improvement by 3 days after a single dose and 4 weeks after multiple dosing. Effects on fertility in males were completely reversed within nine weeks of discontinuation of multiple dosing. Multiple doses of 10 and 100 mg/kg/day tamsulosin hydrochloride (1/5 and 16 times the anticipated human AUC exposure) did not significantly alter fertility in male rats. Effects of tamsulosin hydrochloride on sperm counts or sperm function have not been evaluated.
Studies in female rats revealed significant reductions in fertility after single or multiple dosing with 300 mg/kg/day of the R-isomer or racemic mixture of tamsulosin hydrochloride, respectively. In female rats, the reductions in fertility after single doses were considered to be associated with impairments in fertilization. Multiple dosing with 10 or 100 mg/kg/day of the racemic mixture did not significantly alter fertility in female rats.
14. Clinical Studies
Four placebo-controlled clinical studies and one active-controlled clinical study enrolled a total of 2296 patients (1003 received FLOMAX capsules 0.4 mg once daily, 491 received FLOMAX capsules 0.8 mg once daily, and 802 were control patients) in the U.S. and Europe.
In the two U.S. placebo-controlled, double-blind, 13-week, multicenter studies [Study 1 (US92-03A) and Study 2 (US93-01)], 1486 men with the signs and symptoms of BPH were enrolled. In both studies, patients were randomized to either placebo, FLOMAX capsules 0.4 mg once daily, or FLOMAX capsules 0.8 mg once daily. Patients in FLOMAX capsules 0.8 mg once-daily treatment groups received a dose of 0.4 mg once daily for one week before increasing to the 0.8 mg once-daily dose. The primary efficacy assessments included: 1) total American Urological Association (AUA) Symptom Score questionnaire, which evaluated irritative (frequency, urgency, and nocturia), and obstructive (hesitancy, incomplete emptying, intermittency, and weak stream) symptoms, where a decrease in score is consistent with improvement in symptoms; and 2) peak urine flow rate, where an increased peak urine flow rate value over baseline is consistent with decreased urinary obstruction.
Mean changes from baseline to Week 13 in total AUA Symptom Score were significantly greater for groups treated with FLOMAX capsules 0.4 mg and 0.8 mg once daily compared to placebo in both U.S. studies (Table 3, Figures 2A and 2B). The changes from baseline to Week 13 in peak urine flow rate were also significantly greater for the FLOMAX capsules 0.4 mg and 0.8 mg once-daily groups compared to placebo in Study 1, and for the FLOMAX capsules 0.8 mg once-daily group in Study 2 (Table 3, Figures 3A and 3B). Overall there were no significant differences in improvement observed in total AUA Symptom Scores or peak urine flow rates between the 0.4 mg and the 0.8 mg dose groups with the exception that the 0.8 mg dose in Study 1 had a significantly greater improvement in total AUA Symptom Score compared to the 0.4 mg dose.
Table 3. Mean (±S.D.) Changes from Baseline to Week 13 in Total AUA Symptom Score** and Peak Urine Flow Rate (mL/sec):
| Total AUA Symptom Score | Peak Urine Flow Rate | |||
|---|---|---|---|---|
| Mean Baseline Value | Mean Change | Mean Baseline Value | Mean Change | |
| Study 1† | ||||
| FLOMAX capsules 0.8 mg once daily | 19.9 ± 4.9 n=247 | -9.6* ± 6.7 n=237 | 9.57 ± 2.51 n=247 | 1.78* ± 3.35 n=247 |
| FLOMAX capsules 0.4 mg once daily | 19.8 ± 5.0 n=254 | -8.3* ± 6.5 n=246 | 9.46 ± 2.49 n=254 | 1.75* ± 3.57 n=254 |
| Placebo | 19.6 ± 4.9 n=254 | -5.5 ± 6.6 n=246 | 9.75 ± 2.54 n=254 | 0.52 ± 3.39 n=253 |
| Study 2‡ | ||||
| FLOMAX capsules 0.8 mg once daily | 18.2 ± 5.6 n=244 | -5.8* ± 6.4 n=238 | 9.96 ± 3.16 n=244 | 1.79* ± 3.36 n=237 |
| FLOMAX capsules 0.4 mg once daily | 17.9 ± 5.8 n=248 | -5.1* ± 6.4 n=244 | 9.94 ± 3.14 n=248 | 1.52 ± 3.64 n=244 |
| Placebo | 19.2 ± 6.0 n=239 | -3.6 ± 5.7 n=235 | 9.95 ± 3.12 n=239 | 0.93 ± 3.28 n=235 |
* Statistically significant difference from placebo (p-value ≤0.050; Bonferroni-Holm multiple test procedure).
** Total AUA Symptom Scores ranged from 0 to 35.
† Peak urine flow rate measured 4 to 8 hours post dose at Week 13.
‡ Peak urine flow rate measured 24 to 27 hours post dose at Week 13.
Week 13: For patients not completing the 13-week study, the last observation was carried forward.
Mean total AUA Symptom Scores for both FLOMAX capsules 0.4 mg and 0.8 mg once-daily groups showed a rapid decrease starting at 1 week after dosing and remained decreased through 13 weeks in both studies (Figures 2A and 2B).
In Study 1, 400 patients (53% of the originally randomized group) elected to continue in their originally assigned treatment groups in a double-blind, placebo-controlled, 40-week extension trial (138 patients on 0.4 mg, 135 patients on 0.8 mg, and 127 patients on placebo). Three hundred twenty-three patients (43% of the originally randomized group) completed one year. Of these, 81% (97 patients) on 0.4 mg, 74% (75 patients) on 0.8 mg, and 56% (57 patients) on placebo had a response ≥25% above baseline in total AUA Symptom Score at one year.
Figure 2A. Mean Change from Baseline in Total AUA Symptom Score (0-35) Study 1:
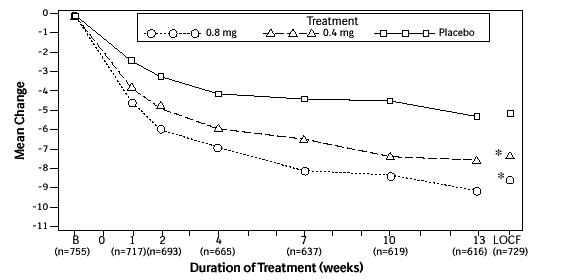
* indicates significant difference from placebo (p-value ≤0.050).
B = Baseline determined approximately one week prior to the initial dose of double-blind medication at Week 0.
Subsequent values are observed cases.
LOCF = Last observation carried forward for patients not completing the 13-week study.
Note: Patients in the 0.8 mg treatment group received 0.4 mg for the first week.
Note: Total AUA Symptom Scores range from 0 to 35.
Figure 2B. Mean Change from Baseline in Total AUA Symptom Score (0-35) Study 2:

* indicates significant difference from placebo (p-value ≤0.050).
Baseline measurement was taken Week 0. Subsequent values are observed cases.
LOCF = Last observation carried forward for patients not completing the 13-week study.
Note: Patients in the 0.8 mg treatment group received 0.4 mg for the first week.
Note: Total AUA Symptom Scores range from 0 to 35.
Figure 3A. Mean Increase in Peak Urine Flow Rate (mL/Sec) Study 1:
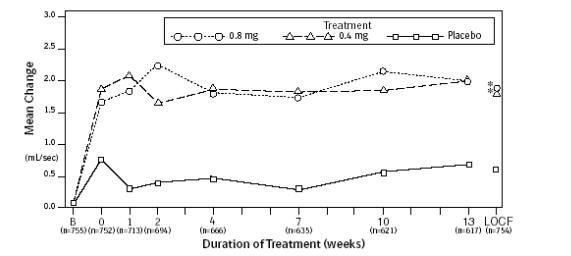
* indicates significant difference from placebo (p-value ≤0.050).
B = Baseline determined approximately one week prior to the initial dose of double-blind medication at Week 0.
Subsequent values are observed cases.
LOCF = Last observation carried forward for patients not completing the 13-week study.
Note: The uroflowmetry assessments at Week 0 were recorded 4 to 8 hours after patients received the first dose of double-blind medication.
Measurements at each visit were scheduled 4 to 8 hours after dosing (approximate peak plasma tamsulosin concentration).
Note: Patients in the 0.8 mg treatment groups received 0.4 mg for the first week.
Figure 3B. Mean Increase in Peak Urine Flow Rate (mL/Sec) Study 2:
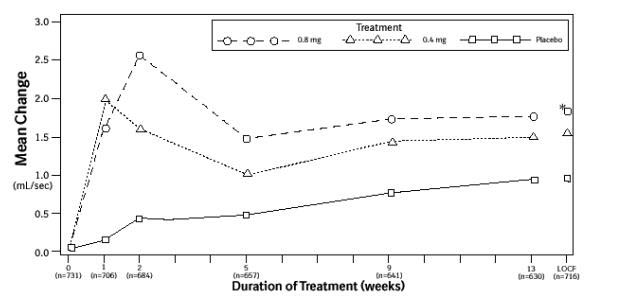
* indicates significant difference from placebo (p-value ≤0.050).
Baseline measurement was taken Week 0. Subsequent values are observed cases.
LOCF = Last observation carried forward for patients not completing the 13-week study.
Note: Patients in the 0.8 mg treatment group received 0.4 mg for the first week.
Note: Week 1 and Week 2 measurements were scheduled 4 to 8 hours after dosing (approximate peak plasma tamsulosin concentration).
All other visits were scheduled 24 to 27 hours after dosing (approximate trough tamsulosin concentration).
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.