FOTIVDA Hard capsule Ref.[7672] Active ingredients: Tivozanib
Source: European Medicines Agency (EU) Revision Year: 2019 Publisher: EUSA Pharma (Netherlands) B.V., Johannes Vermeerplein 11, 1071 DV, Amsterdam, Netherlands
Pharmacodynamic properties
Pharmacotherapeutic group: antineoplastic agents, protein-kinase inhibitors
ATC code: L01XE34
Mechanism of action
Tivozanib potently and selectively blocks all 3 Vascular Endothelial Growth Factor receptors (VEGFR) and has been shown to block various VEGF-induced biochemical and biologic responses in vitro, including VEGF-ligand-induced phosphorylation of all three VEGFR 1, 2 and 3, and proliferation of human endothelial cells. The next most potently inhibited kinase is c-kit which is 8-fold less sensitive to inhibition by tivozanib compared to VEGFR 1, 2 and 3. VEGF is a potent mitogenic factor that plays a central role in angiogenesis and vascular permeability of tumour tissues. By blocking VEGF-induced VEGFR activation, tivozanib inhibits angiogenesis and vascular permeability in tumour tissues, leading to inhibition of tumour growth in vivo.
Clinical efficacy
The efficacy of tivozanib in the treatment of advanced RCC was studied in the following randomised clinical study.
Study AV-951-09-301
This controlled clinical study was a Phase 3 multi-centre, open-label, international, randomised study comparing tivozanib with sorafenib in patients with advanced RCC. Five hundred and seventeen (517) patients with recurrent or metastatic RCC with a clear cell component were randomised (1:1) to receive either tivozanib 1340 microgram once daily on a schedule of 3 weeks on treatment followed by 1 week off (schedule 3/1) or sorafenib 400 mg twice a day. The study included patients who had all undergone prior nephrectomy, and who had received either no prior therapy or no more than one prior systemic therapy in the metastatic setting (immunotherapy/chemotherapy); prior treatment with VEGF or mechanistic Target of Rapamycin (mTOR) targeted therapy was not allowed. Cross-over to the tivozanib arm was permitted upon Response Evaluation Criteria In Solid Tumours (RECIST)-defined progression on sorafenib according to the protocol of a separate extension study.
The primary endpoint of the study was progression-free survival (PFS) by blinded independent radiology review; key secondary endpoints included overall survival (OS) and objective response rate (ORR) by independent radiology review.
The intent-to-treat (ITT) population included 517 patients, 260 randomised to tivozanib and 257 randomised to sorafenib. The baseline demographic and disease characteristics were generally well balanced across the tivozanib and sorafenib arms with regard to age (mean age 58.2 vs 58.4 years respectively), gender (71.2% vs 73.5% male respectively), race (95.8% vs 96.9% white respectively), geographic region (88.1% vs 88.7% from Central/Eastern Europe respectively) and prior treatment for metastatic RCC (69.6% vs 70.8% treatment naïve respectively). For the 30% of patients receiving prior treatment, the predominant therapy was interferon alpha as monotherapy which was received by 75 patients in the tivozanib arm and 62 patients in the sorafenib arm.
Tivozanib showed a statistically significant improvement in PFS and ORR over sorafenib by independent radiology review (Table 2 and Figure 1).
Figure 1. Kaplan-Meier plot of progression free survival, independent radiological review (ITT Population):
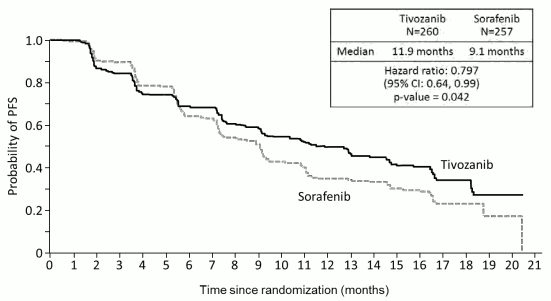
Table 2. Efficacy analysis by independent radiology review (ITT population):
| Tivozanib | Sorafenib | Hazard Ratio (95% CI) | P-value (Log rank test) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Progression-Free Survival [median, months (95% CI)], ITT Population | N=260 | 11.9 (9.3. 14.7) | N=257 | 9.1 (7.3. 9.5) | 0.797 (0.639. 0.993)a | 0.042b | |||||||
| Objective Response Rate (95% CI), ITT Population | N=260 | 33.1% (27.4, 39.2) | N=257 | 23.3% (18.3, 29.0) | 0.014c | Progression-Free Survival, No prior treatment for Metastatic RCC Subgroup [median, months (95% CI)] | N=181 | 12.7 (9.1, 15.0) | N=181 | 9.1 (7.3, 10.8) | 0.756 (0.580, 0.985)d | 0.037e | |
| Progression-Free Survival, One Prior Therapy for Metastatic Disease Subgroup [median, months (95% CI)] | N=78 | 11.9 (8.0, 16.6) | N=76 | 9.1 (7.2, 11.1) | 0.877 (0.587, 1.309)d | 0.520e | |||||||
a Hazard ratio for tivozanib arm vs. sorafenib arm, based on stratified Cox proportional hazards model. Stratification factors are number of prior treatments (0 or 1) and number of metastatic sites/organs involved (1 or ≥2). Assuming proportional hazards, a hazard ratio less than 1 indicates a reduction in hazard rate in favour of tivozanib;
b p-value based on stratified log-rank test. Stratification factors are number of prior treatments (0 or 1) and number of metastatic sites/organs involved (1 or ≥2);
c p-value based on stratified Cochran-Mantel-Haenszel (CMH) statistic. Stratification factors are number of prior treatments (0 or 1) and number of metastatic sites/organs involved (1 or ≥2);
d Hazard ratio for tivozanib arm vs. sorafenib arm subgroup analyses, based on unstratified Cox proportional hazards model. Assuming proportional hazards, a hazard ratio less than 1 indicates a reduction in hazard rate in favour of tivozanib;
e p-value for subgroup analyses based on unstratified log-rank test.
OS was a key secondary endpoint in the pivotal study and the analysis included data from all randomized patients, including those who progressed on sorafenib and crossed over to receive tivozanib as part of the extension study. In the ITT population there was a small numerical difference between the two arms in terms of overall survival. median OS was 28.2 months (95% CI 22.5, 33.0) in the tivozanib arm compared to 30.8 months (95% CI 28.4, 33.3) in the sorafenib arm (HR=1.147, p=0.276).
Elderly patients
In a controlled clinical study (AV-951-09-301), in which 25% of patients receiving tivozanib were ≥65 years of age, no overall differences was observed in efficacy between elderly and younger patients (see section 4.2). In the core RCC studies some adverse reaction occurred more commonly in the elderly (see section 4.4).
Pharmacodynamic effects
In a cardiac safety study of 50 patients with advanced solid tumours treated with tivozanib at 1340 microgram daily for 21 days, the mean change from baseline in QTcF was 6.8 ms on day 21 of dosing. The maximum change in QTcF from baseline was 9.3 ms (90% CI: 5, 13.6), which occurred 2.5 hours after dosing on Day 21. The central tendency change for all measured days and across all time points was 2.2 ms. No subjects had a new >500 ms change in QTcF; 2 patients (4%) had QTcF values >480 ms. One subject (2%) had a >60 ms change from baseline in QTcF and 6 subjects (12%) had a 30 ms to 60 ms change from baseline (see section 4.4 and section 4.8).
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with tivozanib in all subsets of the paediatric population in advanced renal cell carcinoma (see section 4.2 for information on paediatric use).
Pharmacokinetic properties
Absorption
Following oral administration of tivozanib, peak serum levels are achieved after approximately 2 to 24 hours. After a single 1340 microgram dose, mean Cmax was 10.2 to 25.2 ng/mL across healthy subject and patient studies. Single dose AUC0-inf for healthy volunteers dosed with 1340 microgram tivozanib was 1,950 to 2,491 ng.hr/mL. After once daily dosing of 1340 microgram tivozanib for 21 or 28 days in RCC patients, Cmax was 67.5 to 94.3 ng/mL and AUC0-24 was 1,180 to 1,641 ng.hr/mL. Exposure is dose proportional between 890 and 1340 microgram and dose related over the wider range of 450 mg and 1790 microgram. Accumulation at steady-state is approximately 6- to 7- fold the exposure observed at single-dose levels. Clearance is similar between acute and chronic dosing indicating no time dependent changes in PK.
When tivozanib was evaluated in a food effect study in healthy subjects, a high fat meal decreased the peak serum concentrations (Cmax) by 23.4% compared to the fasted state. There was no effect of food on the overall exposure (AUC). Based on these data, tivozanib can be dosed with or without food (see section 4.2).
Distribution
In vitro protein binding studies have shown that tivozanib is >99% bound to plasma proteins. No concentration dependence of plasma protein binding was observed over the range of 0.1 to 5 μmol/L tivozanib. Albumin is the major tivozanib binding component in human plasma. In vitro studies have shown that tivozanib is neither a substrate nor an inhibitor of the multidrug efflux pump, P-glycoprotein. In vitro studies suggest that tivozanib is an inhibitor of intestinal BCRP.
Biotransformation
In vitro metabolism studies have shown that CYP3A4 and CYP1A1 are capable of metabolising tivozanib. Unchanged tivozanib is the major circulating form of the molecule, and there were no major metabolites detected in serum at exposure equal to or greater than 10% of the total radioactivity exposure. As CYP1A1 is primarily expressed in extrahepatic tissues such as the lung and intestine, it was considered unlikely that this isoform would be extensively involved in hepatic metabolism.
In vitro studies have shown that metabolites of tivozanib can undergo UGT mediated biotransformation via the UGT1A1, UGT1A3, UGT1A7, UGT1A8, UGT1A9, and UGT1A10 pathways. Direct N-glucoronidation of tivozanib was a minor pathway of metabolism in vitro.
Elimination
After chronic dosing of tivozanib in RCC patients for 21 days followed by 7 days without administration of tivozanib, tivozanib Cmin is approximately 16.0 to 30.9 ng/mL. In studies that evaluated the terminal elimination phase, tivozanib had a mean t1⁄2 of 4.5-5.1 days. After a single dose oral dose of [14C] tivozanib, approximately 79% of the radioactivity was recovered in the faeces and approximately 12% was found in the urine as metabolites. There was no unchanged tivozanib recovered in the urine indicating that tivozanib does not undergo renal excretion. [14C] Tivozanib was the predominant drug-related material in faeces. There were no [14C]-containing metabolites present in faeces at greater than 10% of the dose.
Special populations
Age, gender and race
Based on the population pharmacokinetic analysis, there is no clinically relevant effect of age, gender or race on the pharmacokinetics of tivozanib. Hepatic impairment Results from a single dose study to evaluate the pharmacokinetics, safety and tolerability of tivozanib in subjects with hepatic impairment show that across the entire measurement period, tivozanib was eliminated more slowly in subjects with moderate (Child-Pugh Class B) or severe (Child-Pugh Class C) hepatic impairment. Tivozanib exposure was increased in patients with severe hepatic impairment (mean AUC0-∞ by 4.0-fold) and in patients with moderate hepatic impairment (mean AUC0-∞ by 2.6-fold). No significant increase in exposure was observed in patients with mild (Child-Pugh Class A) hepatic impairment (mean AUC0-∞ by 1.2-fold). Tivozanib should be used with caution in patients with moderate hepatic impairment and the dose reduced to one 1340 microgram capsule every other day. Tivozanib should not be used in patients with severe hepatic impairment (see section 4.2 and section 4.4).
Renal impairment
Clinical studies with tivozanib were conducted in RCC patients with serum creatinine concentration ≤2 times the upper limit of normal, including those who may have had a prior nephrectomy. Although the impact of further impairment of renal function on the overall disposition of tivozanib is unknown, a clinical study has shown that no unchanged tivozanib is excreted in the urine indicating that tivozanib does not undergo renal excretion.. According to the population pharmacokinetic analysis of tivozanib exposure, no dose adjustment is required in patients with mild or moderate renal impairment. Experience of tivozanib use in patients with severe renal impairment is limited and caution is advised.
CYP and UGT in vitro studies
In vitro studies with tivozanib indicate that it is not a CYP enzyme inducer. In vitro studies conducted in human liver microsomes and hepatocytes evaluating the activity of CYP1A2, CYP2B6, CYP2A6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP3A4 suggested that tivozanib is a weak inhibitor of CYP2B6 and CYP2C8. Based on the in vitro IC 50 and in vivo unbound Cmax, tivozanib was unlikely to interact in a clinically relevant manner with active substances that are metabolised by these enzyme pathways.
Studies conducted in vitro have shown that tivozanib is not a potent inhibitor of UGT (UDP-glucuronosyltransferase) metabolic activities and clinically relevant drug-drug interactions are unlikely with medicinal products metabolised by these pathways.
Transporter in vitro studies
In vitro studies have shown that tivozanib is neither a substrate nor inhibitor of the transporter proteins MDR1 (P-gp), OCT1, OATP1B1, OATP1B3 and BSEP. Furthermore, tivozanib was not an in vitro inhibitor of OAT1, OAT3, OCT2, MATE1 and MATE2-K or substrate of MRP2 and BCRP.
Tivozanib inhibits the transporter protein BCRP in vitro, at concentrations that are likely to restrict the effect to intestinal BCRP activity in vivo.
Preclinical safety data
Adverse reactions not observed in clinical studies, but seen in animals at exposure levels similar to clinical exposure levels and with possible relevance to clinical use were as follows.
In repeat-dose toxicity studies in rats, abnormalities were noted in growing incisors (thin brittle teeth, tooth loss, malocclusions) at doses approximately 2-fold greater than the calculated human equivalent dose and growth plate hypertrophy was observed at doses approximately 0.7- to 7-fold greater than the calculated human equivalent dose. Tivozanib was shown to cause growth plate hypertrophy, absence of active corpora lutea and no maturing follicles in cynomolgus monkeys at dose levels that produced exposures equivalent to those seen at the recommended clinical dose.
Reproduction, mutagenesis, impairment of fertility
Tivozanib may impair human fertility. In nonclinical studies assessing mating and fertility parameters in male rats, doses >2-fold higher than the recommended clinical dose, produced increased epididymis and testis weights associated with infertility. Increased testis weights were observed at a dose 7-fold higher than the recommended clinical dose. In female rats, an increase in non-viable foetuses was noted at a dose 0.7-fold the recommended clinical dose, while dose levels ≥2 fold the recommended clinical dose produced infertility.
Tivozanib was shown to be teratogenic, embryotoxic and foetotoxic in pregnant rats at dose levels 5 times lower than the recommended clinical dose (based on a 60 kg human). Studies in pregnant rabbits showed no effect on maternal health or embryo foetal development at doses approximately 0.6 times the human exposure at the recommended dose.
Carcinogenesis
Carcinogenicity studies have not been performed with tivozanib.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.