Source: European Medicines Agency (EU) Revision Year: 2020 Publisher: Alnylam Netherlands B.V., Strawinskylaan 3051, 1077 ZX Amsterdam, Netherlands
Pharmacotherapeutic group: not yet assigned
ATC code: not yet assigned
Givosiran is a double-stranded small interfering ribonucleic acid (siRNA) that causes degradation of aminolevulinic acid synthase 1 (ALAS1) messenger ribonucleic acid (mRNA) in hepatocytes through RNA interference, resulting in a reduction of induced liver ALAS1 mRNA towards normal. This leads to reduced circulating levels of neurotoxic intermediates aminolevulinic acid (ALA) and porphobilinogen (PBG), the key causal factors of attacks and other disease manifestations of AHP.
In the placebo-controlled study in patients with AHP receiving givosiran 2.5 mg/kg once monthly (ENVISION), median reductions from baseline in urinary ALA and PBG of 83.7 % and 75.1 %, respectively, were observed 14 days after the first dose. Maximal reductions in ALA and PBG levels were achieved around month 3 with median reductions from baseline of 93.8 % for ALA and 94.5 % for PBG, and were sustained with repeated once monthly dosing.
Observed data and modelling demonstrated that once monthly dosing with 2.5 mg/kg givosiran resulted in a greater reduction and less fluctuation in ALA levels compared with doses less than 2.5 mg/kg or dosing once every 3 months.
The efficacy of givosiran was evaluated in a randomised, double-blind, placebo-controlled, multinational study (ENVISION).
A total number of 94 patients with AHP (89 patients with acute intermittent porphyria (AIP), 2 patients with variegate porphyria (VP), 1 patient with hereditary coproporphyria (HCP), and 2 patients with no identified mutation in a porphyria-related gene) were randomised 1:1 to receive once monthly subcutaneous injections of givosiran 2.5 mg/kg or placebo during the 6-month double-blind period. Patients randomised to givosiran included 46 patients with AIP, 1 patient with VP, and 1 patient with HCP. In this study, inclusion criteria specified a minimum of 2 porphyria attacks requiring hospitalisation, urgent healthcare visit, or intravenous (IV) hemin administration at home in the 6 months prior to study entry. Hemin use during the study was permitted for the treatment of acute porphyria attacks. The median age of patients in the ENVISION study was 37.5 years (range 19 to 65 years); 89.4 % of patients were female, and 77.7 % were white. The treatment arms were balanced with respect to historical annualised porphyria attack rate (overall median baseline rate of 8 per year), prior hemin prophylaxis, use of opioid medicinal products, and patient-reported measures of chronic symptoms between attacks.
The major efficacy measure was the annualised attack rate (AAR) of composite porphyria attacks during the 6-month double-blind period and consisted of three components: attacks requiring hospitalisation, urgent healthcare visit, or IV hemin administration at home. This composite efficacy measure was evaluated as the primary endpoint in patients with AIP, and as a secondary endpoint in the overall population of patients with AHP. Treatment with this medicinal product resulted in a significant reduction of the AAR of composite porphyria attacks, compared with placebo, of 74 % in patients with AIP (Table 2). Comparable results were seen in patients with AHP, with a reduction of 73 %. Consistent results were observed for each of the 3 components of the composite porphyria attack endpoint.
The results observed over 6 months were maintained through Month 12, with a median AAR (Q1, Q3) of 0.0 (0.0, 3.5) observed for patients with continued dosing with the medicinal product during the open-label extension period.
Givosiran reduced porphyria attacks compared to placebo in patients with AHP across all pre-specified subgroups, including age, sex, race, region, baseline body mass index (BMI), prior hemin prophylaxis use, historical attack rate, prior chronic opioid use when not having attacks, and the presence of prior chronic symptoms when not having attacks.
Additional clinical efficacy endpoints were studied in AIP patients and are summarised in Table 2.
Table 2. Clinical Efficacy Results in Patients with AIP during the 6-Month Double-Blind Period of the ENVISION Study:
| ΚEndpoint | Placebo (N=43) | Givosiran (N=46) |
|---|---|---|
| Annualised attack rate of composite porphyria attacksa | ||
| Mean AAR (CI 95%)b | 12,5 (9,4, 16,8) | 3,2 (2,3, 4,6) |
| Rate ratio (CI 95%)b (givosiran/placebo) | 0,26 (0,16, 0,41) | |
| P-valueb | <0,001 | |
| Median AAR, (Q1, Q3) | 10,7 (2,2, 26,1) | 1,0 (0,0, 6,2) |
| Number of patients with 0 attacks (%) | 7 (16,3) | 23 (50,0) |
| Annualised days of hemin use | ||
| Mean (CI 95%)b | 29,7 (18,4, 47,9) | 6,8 (4,2, 10,9) |
| Ratio (CI 95%)b (givosiran/placebo) | 0,23 (0,11, 0,45) | |
| P-valueb | <0,001 | |
| Daily worst pain scorec | ||
| Baseline, median (Q1, Q3) | 3,3 (1,9, 5,6) | 2,2 (1,2, 4,5) |
| Median of treatment difference (95 %) (givosiran-placebo) | -10,1 (-22,8, 0,9) | |
| P-value | <0,05 | |
| PCS of SF12d | ||
| Baseline, mean (SD) | 38,4 (9,4) | 39,4 (9,6) |
| Change from baseline at Month 6, LS mean (95 % CI) | 1,4 (−1,0, 3,9) | 5,4 (3,0, 7,7) |
| LS mean difference (95 % CI) (givosiranplacebo) | 3,9 (0,6, 7,3) | |
| Nominal P-value | <0,05 | |
AAR, Annualised Attack Rate; AIP, Acute Intermittent Porphyria; CI, Confidence Interval; Q1, Quartile 1; Q3, Quartile 3; LS, Least Square; PCS, Physical Component Summary; SF-12, the 12-item Short-Form Health Survey
a Composite porphyria attacks includes three components: attacks requiring hospitalisation, urgent healthcare visits, or IV hemin administration at home.
b Based on negative binomial regression model. A rate ratio < 1 represents a favourable outcome for givosiran.
c Patients provided a daily self-assessment of their worst pain based on a 0 to 10 numerical rating scale (NRS). A lower score indicates fewer symptoms. Median of treatment difference and CI were estimated using the Hodges-Lehmann method; p-value was based on Wilcoxon rank sum test, which was conducted post-hoc after data showed a significant deviation from normal distribution.
d A higher score indicates improved health-related quality of life; analysed using the mixed-effect model repeated measures (MMRM) method. The endpoint was not formally tested for statistical significance; a nominal p-value was reported.
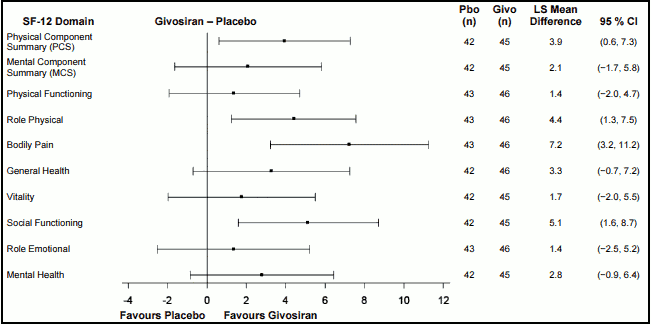
In addition to greater improvement from baseline in the SF-12 PCS score compared to patients treated with placebo at Month 6, there was consistent evidence of effect favouring this medicinal product in bodily pain, role-physical, and social functioning domains, but not in the general health, physical functioning, role-emotional, vitality, and mental health domains (Figure 1).
Figure 1. Change from Baseline to Month 6 in SF-12 Domain Scores in Patients with AIP:
AIP, Acute Intermittent Porphyria; CI, Confidence Interval; Givo, givosiran; Pbo, placebo; LS, Least Square; MCS, Mental Component Summary; PCS, Physical Component Summary; SF-12, the 12-item Short-Form health survey version 2.
In a patient global assessment (Patient Global Impression of Change – PGIC) a larger proportion of patients with AIP treated with givosiran (61.1 %) than with placebo (20 %) rated their overall status as “very much improved” or “much improved” since the start of the study.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with this medicinal product in all subsets of the paediatric population in the treatment of AHP (see section 4.2 and section 5.2 for information on paediatric use).
Following subcutaneous administration, givosiran is rapidly absorbed with a time to maximum plasma concentration (tmax) of 0.5 to 2 hours. At the 2.5 mg/kg once monthly dose, the steady-state peak plasma concentrations of givosiran (Cmax) and area under the curve from time of dosing up to 24 hours after dosing (AUC24) were 321 ± 163 ng/mL and 4130 ± 1780 ng·h/mL, respectively, and corresponding values for the active metabolite were 123 ± 79.0 ng/mL and 1930 ± 1210 ng·h/mL, respectively.
Givosiran is greater than 90% bound to plasma proteins over the concentration range observed in humans at the 2.5 mg/kg once monthly dose. The population estimate for the steady state apparent volume of distribution (Vd/F) for givosiran and for the active metabolite was 10.4 L. Givosiran and its active metabolite distribute primarily to the liver after subcutaneous dosing.
Givosiran is metabolised by nucleases to oligonucleotides of shorter lengths. Active metabolite AS(N-1)3' givosiran (with equal potency as that of givosiran) was a major metabolite in plasma with 45% exposure (AUC0-24) relative to givosiran at the 2.5 mg/kg once monthly dose. In vitro studies indicate that givosiran does not undergo metabolism by CYP450 enzymes.
Givosiran and its active metabolite are eliminated from plasma primarily by metabolism with an estimated terminal half-life of approximately 5 hours. The population estimate for apparent plasma clearance was 36.6 L/h for givosiran and 23.4 L/h for AS(N-1)3' givosiran. After subcutaneous dosing, up to 14 % and 13 % of the administered givosiran dose was recovered in urine as givosiran and its active metabolite, respectively, over 24 hours. The renal clearance ranged from 1.22 to 9.19 L/h for givosiran and 1.40 to 12.34 L/h for the active metabolite.
Givosiran and its active metabolite exhibited linear pharmacokinetics in plasma over the 0.35 to 2.5 mg/kg dose range. At doses greater than 2.5 mg/kg, plasma exposure increased slightly greater than dose-proportionally. Givosiran exhibited time-independent pharmacokinetics with chronic dosing at the recommended dose regimen of 2.5 mg/kg once monthly. There was no accumulation of givosiran or the active metabolite in plasma after repeated once monthly dosing.
Plasma concentrations of givosiran are not reflective of the extent or duration of pharmacodynamic activity. Since givosiran is a liver targeted therapy, concentrations in plasma decline rapidly due to uptake by the liver. In the liver, givosiran exhibits a long half-life leading to extended duration of pharmacodynamic effect maintained over the monthly dosing interval.
No studies have been conducted in patients aged >65 years. Age was not a significant covariate in the pharmacokinetics of givosiran.
In clinical studies there was no difference in the pharmacokinetics or pharmacodynamics of givosiran based on gender or race.
Adult patients with mild hepatic impairment (bilirubin ≤1×ULN and AST >1×ULN, or bilirubin >1×ULN to 1.5×ULN) had comparable plasma exposure of givosiran and its active metabolite and similar pharmacodynamics (percent reduction in urinary ALA and PBG) as patients with normal hepatic function. No studies have been conducted in patients with moderate or severe hepatic impairment (see sections 4.2 and 4.4).
Adult patients with mild renal impairment (eGFR ≥60 to <90 mL/min/1.73 m²), moderate renal impairment (eGFR ≥30 to <60 mL/min/1.73 m²) or severe renal impairment (eGFR ≥15 to <30 mL/min/1.73 m²) had comparable plasma exposure of givosiran and its active metabolite and similar pharmacodynamics (percent reduction in urinary ALA and PBG) as patients with normal renal function (eGFR ≥ to 90 mL/min/1.73 m²). No studies have been conducted in patients with end-stage renal disease or patients with dialysis (see sections 4.2 and 4.4).
Available data suggest that body weight but not age was a significant covariate in the pharmacokinetics of givosiran. At the 2.5 mg/kg dose, a similar exposure is expected in adolescents aged 12 years or older, as in adults with the same body weight.
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, toxicity to reproduction and development. In the repeat-dose toxicity studies conducted in rats and monkeys, the rat was identified as the most sensitive species to givosiran-related effects, with the liver being identified as the primary target organ of toxicity in both the rat and monkey. No adverse findings were associated with chronic, weekly administration of givosiran to rats and monkeys at doses that achieved exposure multiples of 3.5- and 26.3-fold, respectively when compared to exposures achieved in patients receiving the maximum recommended human dose.
Givosiran did not exhibit a genotoxic potential in vitro and in vivo.
Animal studies have not been conducted to evaluate the carcinogenic potential of givosiran.
Embryo-foetal development studies have been performed in rats and rabbits during organogenesis. Givosiran showed marked maternal toxicity in rabbits (including mean maternal body weight loss) and resulted in increased post-implantation loss as a result of increased early resorptions and a low incidence of skeletal variations. These findings are considered an indirect effect, secondary to maternal toxicity. No adverse developmental effects were observed in rats administered the maternally toxic dose of approximately 9 times the normalised maximum recommended human dose.
In a postnatal development study in rats, there was no effect on growth and development of the offspring.
No adverse effects were observed in the fertility of male and female rats when administered with givosiran.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.