HALAVEN Solution for injection Ref.[8567] Active ingredients: Eribulin mesylate
Source: European Medicines Agency (EU) Revision Year: 2019 Publisher: Eisai GmbH, Lyoner Straße 36, 60528 Frankfurt am Main, Germany, e-mail: medinfo_de@eisai.net
Contraindications
- Hypersensitivity to the active substance or to any of the excipients listed in section 6.1.
- Breast-feeding.
Special warnings and precautions for use
Haematology
Myelosuppression is dose dependent and primarily manifested as neutropenia (section 4.8). Monitoring of complete blood counts should be performed on all patients prior to each dose of eribulin. Treatment with eribulin should only be initiated in patients with ANC values ≥1.5 x 109/l and platelets >100 x 109/l.
Febrile neutropenia occurred in <5% of patients treated with eribulin. Patients experiencing febrile neutropenia, severe neutropenia or thrombocytopenia, should be treated according to the recommendations in section 4.2.
Patients with alanine aminotransferase (ALT) or aspartate aminotransferase (AST) >3 x upper limit of normal (ULN) experienced a higher incidence of Grade 4 neutropenia and febrile neutropenia. Although data are limited, patients with bilirubin >1.5 x ULN also have a higher incidence of Grade 4 neutropenia and febrile neutropenia.
Fatal cases of febrile neutropenia, neutropenic sepsis, sepsis and septic shock have been reported.
Severe neutropenia may be managed by the use of granulocyte colony-stimulating factor (G-CSF) or equivalent at the physician's discretion in accordance with relevant guidelines (see section 5.1).
Peripheral neuropathy
Patients should be closely monitored for signs of peripheral motor and sensory neuropathy. The development of severe peripheral neurotoxicity requires a delay or reduction of dose (see section 4.2)
In clinical trials, patients with pre-existing neuropathy greater than Grade 2 were excluded. However, patients with pre-existing neuropathy Grade 1 or 2 were no more likely to develop new or worsening symptoms than those who entered the study without the condition.
QT prolongation
In an uncontrolled open-label ECG study in 26 patients, QT prolongation was observed on Day 8, independent of eribulin concentration, with no QT prolongation observed on Day 1. ECG monitoring is recommended if therapy is initiated in patients with congestive heart failure, bradyarrhythmias or concomittant treatment with medicinal products known to prolong the QT interval, including Class Ia and III antiarrhythmics, and electrolyte abnormalities. Hypokalaemia, hypocalcaemia or hypomagnesaemia should be corrected prior to initiating HALAVEN and these electrolytes should be monitored periodically during therapy. Eribulin should be avoided in patients with congenital long QT syndrome.
Excipients
This medicinal product contains small amounts of ethanol (alcohol), less than 100 mg per dose.
Interaction with other medicinal products and other forms of interaction
Eribulin is mainly (up to 70%) eliminated through biliary excretion. The transport protein involved in this process is unknown. Eribulin is not a substrate of breast cancer resistance protein (BCRP), organic anion (OAT1, OAT3, OATP1B1, OATP1B3), multi-drug resistance-associated protein (MRP2, MRP4) and bile salt export pump (BSEP) transporters.
No drug-drug interactions are expected with CYP3A4 inhibitors and inducers. Eribulin exposure (AUC and Cmax) was unaffected by ketoconazole, a CYP3A4 and P glycoprotein (Pgp) inhibitor, and rifampicin, a CYP3A4 inducer.
Effects of eribulin on the pharmacokinetics of other medicines
In vitro data indicate that eribulin is a mild inhibitor of the important drug metabolising enzyme CYP3A4. No in vivo data are available. Caution and monitoring for adverse events is recommended with concomitant use of substances that have a narrow therapeutic window and that are eliminated mainly via CYP3A4-mediated metabolism (eg alfentanil, cyclosporine, ergotamine, fentanyl, pimozide, quinidine, sirolimus, tacrolimus).
Eribulin does not inhibit the CYP enzymes CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6 or 2E1 at relevant clinical concentrations.
At relevant clinical concentrations, eribulin did not inhibit BCRP, OCT1, OCT2, OAT1, OAT3, OATP1B1 and OATP1B3 transporter-mediated activity.
Fertility, pregnancy and lactation
Pregnancy
There are no data from the use of eribulin in pregnant women. Eribulin is embryotoxic, foetotoxic, and teratogenic in rats. HALAVEN should not be used during pregnancy unless clearly necessary and after a careful consideration of the needs of the mother and the risk to the foetus.
Women of childbearing potential must be advised to avoid becoming pregnant whilst they or their male partner are receiving HALAVEN and have to use effective contraception during and up to 3 months after treatment.
Breast-feeding
It is unknown whether eribulin/metabolites are excreted in human or animal breast milk. A risk to newborns/infants cannot be excluded and therefore HALAVEN must not be used during breast-feeding (see section 4.3).
Fertility
Testicular toxicity has been observed in rats and dogs (see section 5.3). Male patients should seek advice on conservation of sperm prior to treatment because of the possibility of irreversible infertility due to therapy with HALAVEN.
Effects on ability to drive and use machines
HALAVEN may cause adverse reactions such as tiredness and dizziness which may lead to minor or moderate influence on the ability to drive or use machines. Patients should be advised not to drive or use machines if they feel tired or dizzy.
Undesirable effects
Summary of safety profile
The most commonly reported adverse reactions related to HALAVEN, are bone marrow suppression manifested as neutropenia, leucopenia, anaemia, thrombocytopenia with associated infections. New onset or worsening of pre-existing peripheral neuropathy has also been reported. Gastrointestinal toxicities, manifested as anorexia, nausea, vomiting, diarrhoea, constipation, and stomatitis are among reported undesirable effects. Other undesirable effects include fatigue, alopecia, increased liver enzymes, sepsis and musculoskeletal pain syndrome.
Tabulated list of adverse reactions
Unless otherwise noted, the table shows the incidence rates of adverse reactions observed in breast cancer and soft tissue sarcoma patients who received the recommended dose in Phase 2 and Phase 3 studies.
Frequency categories are defined as: very common (≥1/10), common (≥1/100 to <1/10), uncommon (≥1/1,000 to <1/100), rare (≥1/10,000 to <1/1,000) and very rare (<1/10,000). Within each frequency grouping, undesirable effects are presented in order of decreasing frequency. Where Grade 3 or 4 reactions occurred, the actual total frequency and the frequency of Grade 3 or 4 reactions are given.
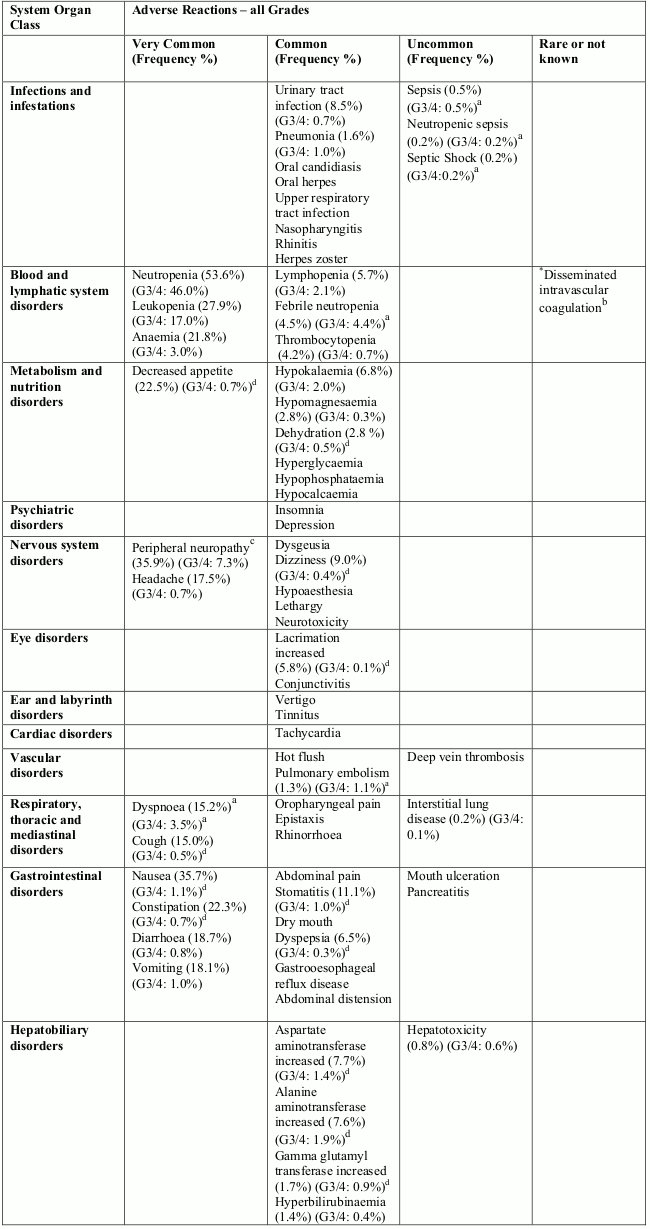
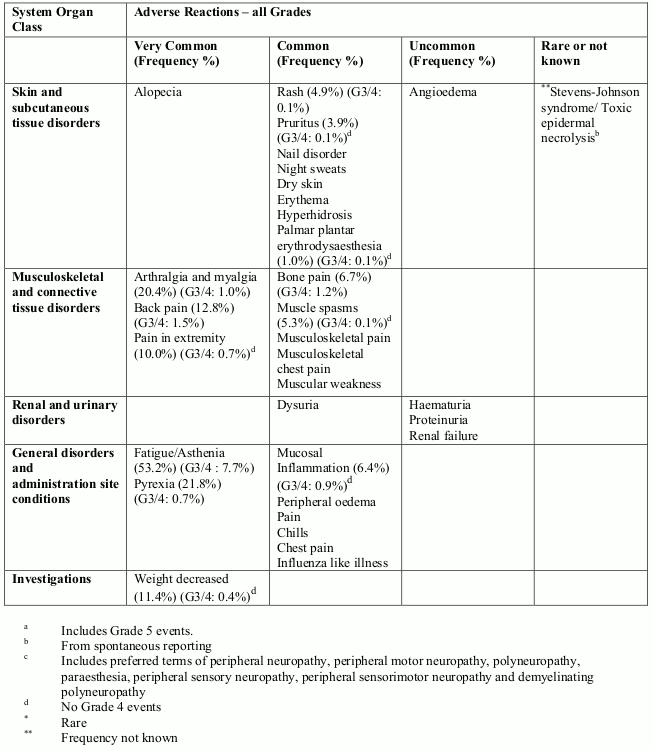
Overall, the safety profiles in the breast cancer and soft tissue sarcoma patient populations were similar.
Description of selected adverse reactions
Neutropenia
The neutropenia observed was reversible and not cumulative; the mean time to nadir was 13 days and the mean time to recovery from severe neutropenia (<0.5 x 109/l) was 8 days.
Neutrophil counts of <0.5 x 109/l that lasted for more than 7 days occurred in 13% of breast cancer patients treated with eribulin in the EMBRACE study.
Neutropenia was reported as a Treatment Emergent Adverse Event (TEAE) in 151/404 (37.4% for all grades) in the sarcoma population, compared with 902/1559 (57.9% for all grades) in the breast cancer population. The combined grouped TEAE and neutrophil laboratory abnormality frequencies were 307/404 (76.0%) and 1314/1559 (84.3%), respectively. The median duration of treatment was 12.0 weeks for sarcoma patients and 15.9 weeks for breast cancer patients.
Fatal cases of febrile neutropenia, neutropenic sepsis, sepsis and septic shock have been reported. Out of 1963 breast cancer and soft tissue sarcoma patients who received eribulin at the recommended dose in clinical trials there was one fatal event each of neutropenic sepsis (0.1%) and febrile neutropenia (0.1%). In addition there were 3 fatal events of sepsis (0.2%) and one of septic shock (0.1%).
Severe neutropenia may be managed by the use of G-CSF or equivalent at the physician's discretion in accordance with relevant guidelines. 18% and 13% of eribulin treated patients received G-CSF in the two phase 3 breast cancer studies (Studies 305 and 301, respectively). In the phase 3 sarcoma study (Study 309), 26% of the eribulin treated patients received G-CSF. Neutropenia resulted in discontinuation in <1% of patients receiving eribulin.
Disseminated intravascular coagulation
Cases of disseminated intravascular coagulation have been reported, typically in association with neutropenia and/or sepsis.
Peripheral neuropathy
In the 1559 breast cancer patients the most common adverse reaction resulting in discontinuation of treatment with eribulin was peripheral neuropathy (3.4%). The median time to Grade 2 peripheral neuropathy was 12.6 weeks (post 4 cycles). Out of the 404 sarcoma patients, 2 patients discontinued treatment with eribulin due to peripheral neuropathy. The median time to Grade 2 peripheral neuropathy was 18.4 weeks.
Development of Grade 3 or 4 peripheral neuropathy occurred in 7.4% of breast cancer patients and 3.5% of sarcoma patients. In clinical trials, patients with pre-existing neuropathy were as likely to develop new or worsening symptoms as those who entered the study without the condition. In breast cancer patients with pre-existing Grade 1 or 2 peripheral neuropathy the frequency of treatment-emergent Grade 3 peripheral neuropathy was 14%.
Hepatotoxicity
In some patients with normal/abnormal liver enzymes prior treatment with eribulin, increased levels of liver enzymes have been reported with initiation of eribulin treatment. Such elevations appeared to have occurred early with eribulin treatment in cycle 1–2 for the majority of these patients and whilst thought likely to be a phenomenon of adaptation to eribulin treatment by the liver and not a sign of significant liver toxicity in most patients, hepatotoxicity has also been reported.
Special populations
Elderly population
Of the 1559 breast cancer patients treated with the recommended dose of eribulin, 283 patients (18.2%) were ≥65 years of age. In the 404 sarcoma patient population, 90 patients (22.3%) treated with eribulin were ≥65 years of age. The safety profile of eribulin in elderly patients (≥65 years of age) was similar to that of patients <65 years of age except for asthenia/fatigue which showed an increasing trend with age. No dose adjustments are recommended for the elderly population.
Patients with hepatic impairment
Patients with ALT or AST >3 x ULN experienced a higher incidence of Grade 4 neutropenia and febrile neutropenia. Although data are limited, patients with bilirubin >1.5 x ULN also have a higher incidence of Grade 4 neutropenia and febrile neutropenia (see also sections 4.2 and 5.2).
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed in Appendix V.
Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal products except those mentioned in section 6.6.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.