HIZENTRA Solution for injection Ref.[27859] Active ingredients: Human normal immunoglobulin G
Source: European Medicines Agency (EU) Revision Year: 2022 Publisher: CSL Behring GmbH, Emil-von-Behring-Strasse 76, D-35041 Marburg, Germany
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: immune sera and immunoglobulins: immunoglobulins, normal human, for extravascular administration
ATC code: J06BA01
Human normal immunoglobulin contains mainly immunoglobulin G (IgG) with a broad spectrum of antibodies against infectious agents.
Human normal immunoglobulin contains the IgG antibodies present in the normal population. It is usually prepared from pooled plasma from not fewer than 1,000 donors. It has a distribution of immunoglobulin G subclasses closely proportional to that in native human plasma.
Mechanism of action
In immunodeficiency, adequate doses of Hizentra may restore abnormally low immunoglobulin G antibody levels to the normal range and thus help against infections. The mechanism of action in indications other than replacement therapy is not fully elucidated, but includes immunomodulatory effects.
PID
In the European pivotal prospective open label, single arm and multicentre study, a total of 51 subjects with primary immunodeficiency syndromes aged between 3 and 60 years old were treated with Hizentra for up to 41 weeks. The mean dose administered each week was 0.12 g/kg body weight (bw). Sustained IgG trough levels with mean concentrations of 7.99–8.25 g/l were thereby achieved throughout the treatment period. Subjects received in total 1,831 weekly Hizentra infusions.
In the US prospective open label, single arm and multicentre study, a total of 49 subjects with primary immunodeficiency syndromes aged between 5 and 72 years old were treated with Hizentra for up to 15 months. The mean dose administered each week was 0.23 g/kg bw. Sustained IgG trough levels with a mean concentration of 12.53 g/l were thereby achieved throughout the treatment period. Subjects received in total 2,264 weekly Hizentra infusions.
No serious bacterial infections were reported during the efficacy period in subjects receiving Hizentra during clinical studies.
To assess the safety and tolerability of higher infusion rates applied via the manual push and pumpassisted administration, 49 PID subjects aged 2 to 75 years were enrolled in an open-label, multicentre, parallel-arm, nonrandomised phase IV HILO (Hizentra Label Optimization) study and treated with Hizentra for at least 12 weeks (11 paediatric patients aged 2 to <18, 35 adult patients aged 18 to 65, and 3 geriatric patients aged >65 years). In the first patient group receiving Hizentra via the manual push technique (n=16), 2 to 7 infusions per week were administered with the flow rates of 30, 60 and 120 ml/hour/site (see section 4.2). In the second patient group receiving Hizentra via pump-assisted administration (n=18), weekly Hizentra infusions were administered with 25, 50, 75 and 100 ml/hour/site flow rate. In a third group, infusion volumes of 25, 40 and 50ml per site were additionally evaluated in pump-assisted administration of weekly Hizentra doses (n=15). In all three groups, each infusion parameter was used for 4 weeks, after which subjects successfully completing required minimal number of valid infusions could switch to the next higher infusion parameter.
The primary endpoint was the percentage of subjects responding to a higher infusion parameter:
| Group | Infusion parameter and responder rate (%) | |||
|---|---|---|---|---|
| 1. manual push flow rates | 30 ml/hour/site | 60 ml/hour/site | 120 ml/hour/site | - |
| 100.0% | 100.0% | 87.5% | - | |
| 2. pump-assisted flow rates | 25 ml/hour/site | 50 ml/hour/site | 75 ml/hour/site | 100 ml/hour/site |
| 77.8% | 77.8% | 66.7% | 61.1% | |
| 3. pump-assisted volumes | 25 ml/site | 40 ml/site | 50 ml/site | - |
| 86.7% | 73.3% | 73.3% | - | |
Responder: in the pump-assisted group a subject who performed ≥3 valid infusions out of 4 for an infusion parameter; in the manual push group a subject who performed ≥60% of valid infusions for an infusion parameter. An infusion was considered valid, if ≥95% of the planned flow rate/volume per ≥1 infusion site was achieved.
Overall, the number of infusions without severe local reactions versus the total number of infusions (tolerability) was ≥0.98 in all groups for all infusion parameters. No clinically relevant differences in the serum IgG trough concentrations were observed between the baseline at day 1 and the end of the study in all subjects.
CIDP
The safety, efficacy and tolerability of Hizentra in patients with CIDP has been assessed in a multicentre, double-blind, randomised, placebo-controlled, parallel-group phase III PATH [Polyneuropathy and Treatment with Hizentra] study. 172 adults with definite or probable CIDP who were previously treated with and responded to IVIg were randomised to weekly 0.2 g/kg bw Hizentra, weekly 0.4 g/kg bw Hizentra or placebo groups, and followed for a subsequent 24 weeks. The mean duration of exposure was 118.9 days in the 0.2 g/kg bw and 129 days in the 0.4 g/kg bw Hizentra group (maximum exposure up to 167 and 166 days in each group, respectively). Subjects generally used 4 infusion sites in parallel (up to 8 sites in parallel). In total, 57 subjects received 1514 infusions in the placebo group, 57 subjects received 2007 infusions in the 0.2 g/kg bw Hizentra group, and 58 subjects received 2218 infusions in the 0.4 g/kg bw Hizentra group (in total 5739 infusions).
The primary efficacy endpoint was the percentage of subjects who had a CIDP relapse (defined as a ≥1 point increase in adjusted Inflammatory Neuropathy Cause and Treatment [INCAT] score compared with baseline) or were withdrawn for any other reason in the Hizentra treatment period. Both Hizentra doses demonstrated superiority over placebo for the primary endpoint. A statistically significant lower percentage of subjects treated with Hizentra, 32.8% for 0.4 g/kg bw and 38.6% for 0.2 g/kg bw, had CIDP relapse or was withdrawn for other reasons compared with 63.2% subjects treated with placebo (p<0.001 or p=0.007, respectively). When only considering relapse, the CIDP relapse rates were 19.0% for 0.4 g/kg bw Hizentra and 33.3% for 0.2 g/kg bw Hizentra compared with 56.1% for placebo (p<0.001 or p=0.012, respectively). Accordingly, over the treatment period for up to 24 weeks Hizentra prevented relapse in 81% and 67% of subjects in the 0.4 g/kg bw and 0.2 g/kg bw group, respectively, while in the placebo group 44% of subjects remained relapse-free.
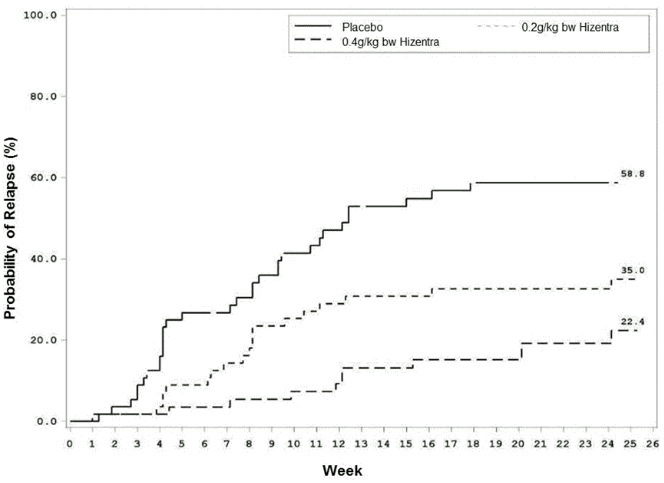
Time to CIDP relapse (Figure 1) was evaluated, and the corresponding probabilities for CIDP relapse based on Kaplan-Meier estimates were: placebo, 58.8%; 0.2 /kg bw Hizentra, 35.0%; and 0.4 g/kg bw Hizentra, 22.4%. The hazard ratios (95% CI) for the lower dose and higher dose compared to placebo was 0.48 (0.27, 0.85) and 0.25 (0.12, 0.49), respectively. The difference observed between the 0.2 g/kg bw and the 0.4 g/kg bw Hizentra groups did not reach statistical significance.
Figure 1. Kaplan-Meier Plot Time to CIDP Relapse:
In the efficacy scores (INCAT score, mean grip strength, and Medical Research Council sum score), subjects in both Hizentra dose groups remained stable while subjects in the placebo group deteriorated. Subjects in the high dose Hizentra group remained stable in the Rasch-built Overall Disability Scale (R-ODS) centile score. Subjects in both Hizentra dose groups remained stable in electrophysiology parameters.
A phase III, multicentre, 48-week open-label extension study enrolled 82 CIDP patients from the PATH study. The extension study investigated the long-term safety and efficacy of Hizentra maintenance therapy in the two weekly doses, 0.2 g/kg and 0.4 g/kg bw. Due to the study design, the same subject could receive both doses during the study; 72 subjects received doses of 0.4 g/kg and 73 subjects received doses of 0.2 g/kg during the efficacy evaluation period. The mean efficacy evaluation period was 125.8 days (range: 1-330) in the 0.2 g/kg, and 196.1 days (range: 1-330) in the 0.4 g/kg bw group. Patients who completed the pivotal PATH study without relapse on 0.4 g/kg bw dose and initially received this dose in the extension study had a relapse rate of 5.6% (1/18 patients). For all patients who received 0.4 g/kg bw in the PATH extension study, 9.7% (7/72 patients) had a relapse. Patients who completed the PATH study without relapse on 0.2 g/kg bw dose and initially received this dose in the extension study had a relapse rate of 50% (3/6 patients). For all patients who received 0.2 g/kg bw in the extension study, 47.9% (35/73 patients) had a relapse. Down-titrating patients in the extension study who completed the PATH study on either dose from 0.4 g/kg to 0.2 g/kg bw dose was possible in 67.9% of subjects (19/28 patients) without occurrence of relapse; all of the 9 relapsers recovered within 4 weeks after treatment with 0.4 g/kg bw dose. Grip strength, MRC sum score, and R-ODS centile score remained stable as compared to baseline for patients who never had a relapse in the extension study.
Paediatric population
The safety and effectiveness of Hizentra have been established in paediatric subjects 2 to 18 years of age. Hizentra was evaluated in 68 paediatric subjects with PID 2 to <12 years of age and in 57 paediatric subjects 12 to <18 years of age. There were no differences in the pharmacokinetics, safety and efficacy profiles as compared with adult subjects. No paediatric-specific dose adjustments were necessary to achieve the desired serum IgG levels. No differences were seen in the pharmacodynamic properties between adult and paediatric study patients with PID. Hizentra has not been evaluated in clinical studies in paediatric patients with CIDP who are under the age of 18.
Elderly
No overall differences in safety or efficacy were observed between PID subjects >65 years and PID subjects 18 to 65 years of age. In the clinical studies Hizentra was evaluated in 13 patients with PID >65 years of age.
No overall differences in safety or efficacy were observed between CIDP subjects >65 years and CIDP subjects 18 to 65 years of age. In the clinical studies with CIDP patients, 61 subjects >65 years of age were treated with Hizentra.
5.2. Pharmacokinetic properties
Absorption and Distribution
Following subcutaneous administration of Hizentra, peak serum levels are achieved after approximately 2 days.
Elimination
IgG and IgG-complexes are broken down in cells of the reticuloendothelial system.
PID
In a clinical phase III trial with Hizentra (n=46), the subjects achieved sustained trough levels (median 8.1 g/l) over a period of 29 weeks when receiving median weekly doses of 0.06 to 0.24 g/kgbw.
Simulations by empirical Population Pharmacokinetic models suggested that comparable IgG exposure levels (AUC0-14days, Cmin,14 days) may be obtained if Hizentra is administered subcutaneously every two weeks using double the weekly dose during maintenance therapy.
These simulations further suggested that comparable serum IgG trough levels can be achieved when the weekly maintenance dose of Hizentra is administered in proportional amounts more frequently than once a week (e.g. 2 times per week, 3 times per week, 5 times per week or daily).
Simulation of 2-3 missed daily doses resulted in a median serum IgG level decrease of ≤4% compared to consistent daily dosing. By replacing the missed doses when daily dosing was resumed, the median concentration profile recovered within 2 to 3 days. However, if missed doses were not replaced when dosing was resumed, it took up to 5-6 weeks for the IgG trough levels to return to steady-state.
Paediatric population
No differences were seen in the pharmacokinetic parameters between adult and paediatric PID study patients.
Elderly
No overall differences in the pharmacokinetic parameters were observed between PID subjects >65 years and subjects 18 to 65 years of age.
CIDP
In the PATH study, subjects (n=172) achieved sustained trough levels over a period of 24 weeks when receiving weekly doses of 0.2 g/kg bw and 0.4 g/kg bw, respectively. The mean (SD) IgG trough concentration after Hizentra treatment in the 0.4 g/kg bw group was 20.4 (3.24) g/l and 15.4 (3.06) g/l in the 0.2 g/kg bw group. Simulations with population-pharmacokinetic models in the PATH study suggest that a comparable IgG exposure (Cmax, AUC0-14days, Cmin,14 days) is achieved when the double weekly Hizentra dose is administered every two weeks in the CIDP subjects. These simulations further suggest that a comparable IgG exposure is correspondingly achieved when the weekly maintenance dose of Hizentra is divided in several, more frequent doses (2 to 7 times per week) in the CIDP patients' population.
Paediatric population
Hizentra has not been evaluated in clinical studies in paediatric patients with CIDP who are under the age of 18.
Elderly
No overall differences in the pharmacokinetic parameters were observed between CIDP subjects >65 years and subjects 18 to 65 years of age.
5.3. Preclinical safety data
Immunoglobulins are a normal constituent of the human body. L-proline is a physiological, nonessential amino acid.
The safety of Hizentra has been assessed in several preclinical studies, with particular reference to the excipient L-proline. Non-clinical data reveal no special risk for humans based on safety pharmacology and toxicity studies.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.