Source: European Medicines Agency (EU) Revision Year: 2020 Publisher: AstraZeneca AB, SE-151 85 Södertälje, Sweden
Pharmacotherapeutic group: Antineoplastic agents, monoclonal antibodies
ATC code: L01XC28
Expression of programmed cell death ligand-1 (PD-L1) protein is an adaptive immune response that helps tumours evade detection and elimination by the immune system. PD-L1 can be induced by inflammatory signals (e.g. IFN-gamma) and can be expressed on both tumour cells and tumour-associated immune cells in tumour microenvironment. PD-L1 blocks T-cell function and activation through interaction with PD-1 and CD80 (B7.1). By binding to its receptors, PD-L1 reduces cytotoxic T-cell activity, proliferation and cytokine production.
Durvalumab is a fully human, immunoglobulin G1 kappa (IgG1κ) monoclonal antibody that selectively blocks the interaction of PD-L1 with PD-1 and CD80 (B7.1). Durvalumab does not induce antibody dependent cell-mediated cytotoxicity (ADCC). Selective blockade of PD-L1/PD-1 and PD-L1/CD80 interactions enhances antitumour immune responses and increases T-cell activation.
The efficacy of IMFINZI was evaluated in the PACIFIC Study, a randomised, double-blind, placebo-controlled, multicentre study in 713 patients with locally advanced, unresectable NSCLC. Patients had completed at least 2 cycles of definitive platinum-based chemotherapy with radiation therapy within 1 to 42 days prior to initiation of the study and had a ECOG performance status of 0 or 1. Ninety-two percent of patients had received a total dose of 54 to 66 Gy of radiation. The study excluded patients who had progressed following chemoradiation therapy, patients with prior exposure to any anti-PD-1 or anti-PD-L1 antibody, patients with active or prior documented autoimmune disease within 2 years of initiation of the study; a history of immunodeficiency; a history of severe immune-mediated adverse reactions; medical conditions that required systemic immunosuppression, except physiological dose of systemic corticosteroids; active tuberculosis or hepatitis B or C or HIV infection or patients receiving live attenuated vaccine within 30 days before or after the start of IMFINZI. Patients were randomised 2:1 to receive 10 mg/kg IMFINZI (n=476) or 10 mg/kg placebo (n=237) via intravenous infusion every 2 weeks for up to 12 months or until unacceptable toxicity or confirmed disease progression. Randomisation was stratified by gender, age (<65 years vs. ≥65 years) and smoking status (smoker vs. non-smoker). Patients with disease control at 12 months were given the option to be re-treated upon disease progression. Tumour assessments were conducted every 8 weeks for the first 12 months and then every 12 weeks thereafter.
Patients were enrolled regardless of their tumour PD-L1 expression level. Where available, archival tumour tissue specimens taken prior to chemoradiation therapy were retrospectively tested for PD-L1 expression on tumour cells (TC) using the VENTANA PD-L1 (SP263) IHC assay. Of the 713 patients randomised, 63% of patients provided a tissue sample of sufficient quality and quantity to determine PD-L1 expression and 37% were unknown.
The demographics and baseline disease characteristics were well balanced between study arms. Baseline demographics of the overall study population were as follows: male (70%), age ≥65 years (45%), age ≥75 years (8%), White (69%), Asian (27%), other (4%), current smoker (16%), past-smoker (75%), never smoker (9%), ECOG Performance Status 0 (49%), ECOG Performance Status 1 (51%). Disease characteristics were as follows: Stage IIIA (53%), Stage IIIB (45%), histological sub-groups of squamous (46%), non-squamous (54%). Of 451 patients with PD L1 expression available, 67% were TC ≥1% [PD-L1 TC 1-24% (32%), PD L1 TC ≥25% (35%)] and 33% were TC <1%.
The two primary endpoints of the study were progression-free survival (PFS) and overall survival (OS) of IMFINZI vs. placebo. Secondary efficacy endpoints included PFS at 12 months (PFS 12) and 18 months (PFS 18) from randomisation and Time from Randomisation to Second Progression (PFS2). PFS was assessed by Blinded Independent Central Review (BICR) according to RECIST v1.1.
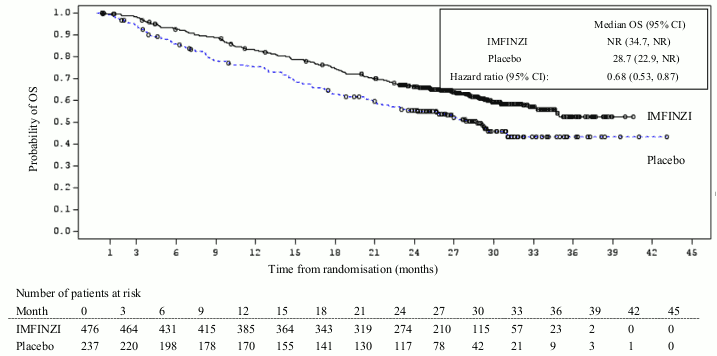
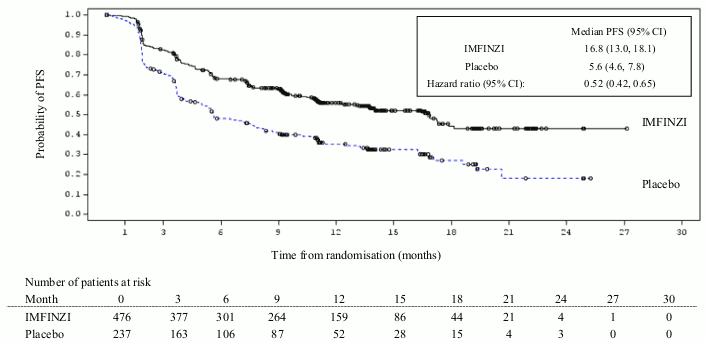
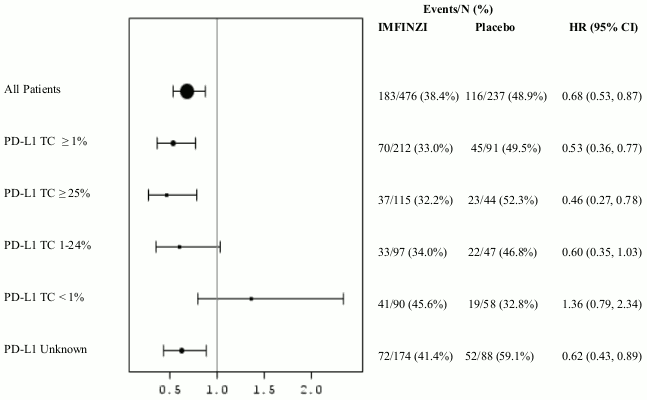
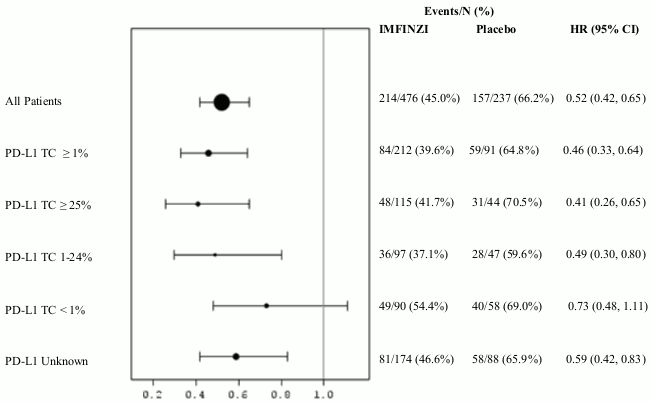
The study demonstrated a statistically significant improvement in PFS in the IMFINZI-treated group compared with the placebo group [hazard ratio (HR)=0.52 (95% CI: 0.42, 0.65), p<0.0001]. The study demonstrated a statistically significant improvement in OS in the IMFINZI-treated group compared with the placebo group [HR=0.68 (95% CI: 0.53, 0.87), p=0.00251]. See Table 3 and Figures 1 and 2.
Table 3. Efficacy results for the PACIFIC Studya:
| IMFINZI (n=476) | Placebo (n=237) | |
|---|---|---|
| OS | ||
| Number of deaths (%) | 183 (38.4%) | 116 (48.9%) |
| Median (months) (95% CI) | NR (34.7, NR) | 28.7 (22.9, NR) |
| HR (95% CI) | 0.68 (0.53, 0.87) | |
| 2-sided | p-value 0.00251 | |
| OS at 24 months () (95 CI) | 66.3% (61.7%, 70.4%) | 55.6% (48.9%, 61.3%) |
| p-value | 0.005 | |
| PFS | ||
| Number of events (%) | 214 (45.0%) | 157 (66.2%) |
| Median PFS (months) (95% CI) | 16.8 (13.0, 18.1) | 5.6 (4.6, 7.8) |
| HR (95% CI) | 0.52 (0.42, 0.65) | |
| p-value | p<0.0001 | |
| PFS at 12 months () (95 CI) | 55.9% (51.0%, 60.4%) | 35.3% (29.0%, 41.7%) |
| PFS at 18 months () (95 CI) | 44.2% (37.7%, 50.5%) | 27.0% (19.9%, 34.5%) |
| PFS2 | ||
| Median PFS2b (months) (95% CI) | 28.3 (25.1, 34.7) | 17.1 (14.5, 20.7) |
| HR (95% CI) | 0.58 (0.46, 0.73) | |
| p-value | p<0.0001 | |
a The analysis of OS was performed approximately 13 months after the primary analysis of PFS.
b PFS2 is defined as the time from the date of randomisation until the date of second progression (defined by local standard clinical practice) or death.
NR: Not Reached
Figure 1. Kaplan-Meier curve of OS:
Figure 2. Kaplan-Meier curve of PFS:
The improvements in PFS and OS in favour of patients receiving IMFINZI compared to those receiving placebo were consistently observed in all predefined subgroups analysed, including ethnicity, age, gender, smoking history, EGFR mutation status and histology.
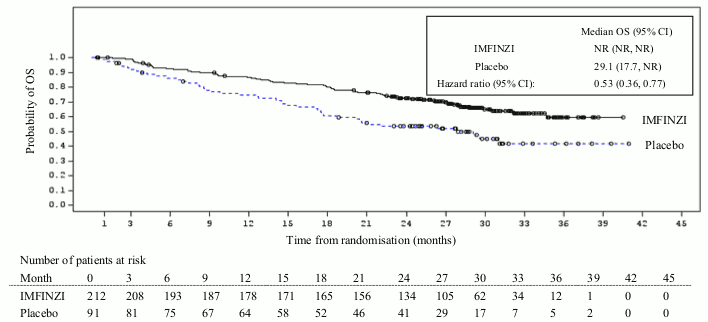
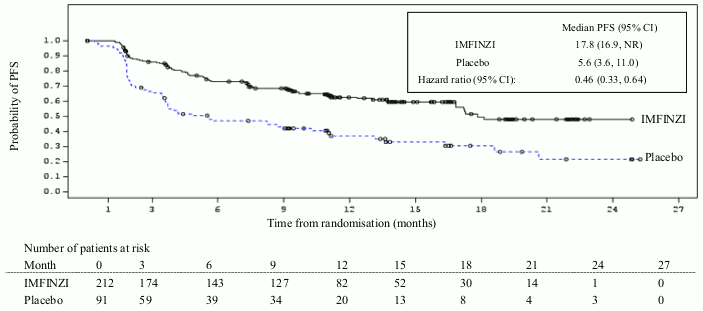
Additional subgroup analyses were conducted to evaluate the efficacy by tumour PD-L1 expression (≥25%, 1-24%, ≥1%, <1%) and for patients whose PD-L1 status cannot be established (PD-L1 unknown). PFS and OS results are summarised in Figures 3, 4, 5 and 6.
Figure 3. Kaplan-Meier curve of OS for PD-L1 TC ≥1%:
Figure 4. Kaplan-Meier curve of PFS for PD-L1 TC ≥1%:
Figure 5. Forest plot of OS by PD-L1 expression:
Figure 6. Forest plot of PFS by PD-L1 expression:
Overall the safety profile of durvalumab in PD-L1 TC ≥1% subgroup was consistent with the intent to treat population, as was the PD-L1 TC <1% subgroup.
Patient-reported symptoms, function and health-related quality of life (HRQoL) were collected using the EORTC QLQ-C30 and its lung cancer module (EORTC QLQ-LC13). The LC13 and C30 were assessed at baseline, every 4 weeks for the first 8 weeks, followed by every 8 weeks until completion of the treatment period or discontinuation of IMFINZI due to toxicity or disease progression. Compliance was similar between the IMFINZI and placebo treatment groups (83% vs 85.1% overall of evaluable forms completed).
At baseline, no differences in patient reported symptoms, function and HRQoL were observed between IMFINZI and placebo groups. Throughout the duration of the study to Week 48, there was no clinically meaningful difference between IMFINZI and placebo groups in symptoms, functioning and HRQoL (as assessed by a difference of greater than or equal to 10 points).
The European Medicines Agency has deferred the obligation to submit the results of studies with durvalumab in all subsets of the paediatric population in the treatment of malignant neoplasms (except central nervous system tumours, haematopoietic and lymphoid tissue neoplasms) (see section 4.2 for information on paediatric use).
The PK of durvalumab was studied in 1902 patients with solid tumours with doses ranging from 0.1 to 20 mg/kg administered intravenously once every two, three or four weeks. PK exposure increased more than dose-proportionally (non-linear PK) at doses <3 mg/kg, and dose proportionally (linear PK) at doses ≥3 mg/kg. Steady state was achieved at approximately 16 weeks. Based on population PK analysis that included 1878 patients in the dose range of ≥10 mg/kg every 2 weeks, the geometric mean steady state volume of distribution (Vss) was 5.64 L. Durvalumab clearance (CL) decreased over time resulting in a geometric mean steady state clearance (CLss) of 8.16 mL/h at Day 365; the decrease in CLss was not considered clinically relevant. The terminal half-life (t1/2), based on baseline CL, was approximately 18 days. The primary elimination pathways of durvalumab are protein catabolism via reticuloendothelial system or target mediated disposition.
Age (19–96 years), body weight (34-149 kg), gender, positive anti-drug antibody (ADA) status, albumin levels, LDH levels, creatinine levels, soluble PD-L1, tumour type, race or ECOG status had no clinically significant effect on the PK of durvalumab.
Mild (creatinine clearance (CrCL) 60 to 89 mL/min) and moderate renal impairment (creatinine clearance (CrCL) 30 to 59 mL/min) had no clinically significant effect on the PK of durvalumab.The effect of severe renal impairment (CrCL 15 to 29 mL/min) on the PK of durvalumab is unknown.
Mild hepatic impairment (bilirubin ≤ ULN and AST > ULN or bilirubin >1.0 to 1.5 × ULN and any AST) had no clinically significant effect on the PK of durvalumab. The effect of moderate hepatic impairment (bilirubin >1.5 to 3 x ULN and any AST) or severe hepatic impairment (bilirubin >3.0 x ULN and any AST) on the pharmacokinetics of durvalumab is unknown; however, as IgG monoclonal antibodies are not primarily cleared via hepatic pathways, a change in hepatic function is not expected to influence durvalumab exposure.
The carcinogenic and genotoxic potential of durvalumab has not been evaluated.
As reported in the literature, the PD-1/PD-L1 pathway plays a central role in preserving pregnancy by maintaining maternal immune tolerance to the foetus, and in mouse allogeneic pregnancy models disruption of PD-L1 signalling was shown to result in an increase in foetal loss. In animal reproduction studies, administration of durvalumab to pregnant cynomolgus monkeys from the confirmation of pregnancy through delivery, at exposure levels approximately 18 times higher than those observed at the clinical dose of 10 mg/kg of durvalumab (based on AUC), was associated with placental transfer but not with maternal toxicity or effects on embryofoetal development, pregnancy outcome or postnatal development. Negligible levels of durvalumab was found in milk of cynomolgous monkey on Day 28 after birth.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.