Source: European Medicines Agency (EU) Revision Year: 2019 Publisher: Amgen Europe B.V., Minervum 7061, NL-4817 ZK Breda, The Netherlands
Pharmacotherapeutic group: Antineoplastic and immunomodulating agents
ATC code: L01XX51
Talimogene laherparepvec is an oncolytic immunotherapy that is derived from HSV-1. Talimogene laherparepvec has been modified to replicate within tumours and to produce the immune stimulatory protein human GM-CSF. Talimogene laherparepvec causes the death of tumour cells and the release of tumour-derived antigens. It is thought that together with GM-CSF, it will promote a systemic anti-tumour immune response and an effector T-cell response. Mice that had complete regression of their primary tumours following treatment were resistant to subsequent tumour rechallenge.
The modifications to talimogene laherparepvec from HSV-1 include deletion of ICP34.5 and ICP47. Whereas anti-viral immune responses defend normal cells following infection by talimogene laherparepvec, tumours have been shown to be susceptible to injury and cell death from ICP34.5-deficient HSV-1 viruses, including talimogene laherparepvec. Deletion of ICP47 prevents down-regulation of antigen presentation molecules and increases the expression of HSV US11 gene, thereby enhancing viral replication in tumour cells.
The safety and efficacy of Imlygic monotherapy compared with subcutaneously administered GM-CSF were evaluated in a phase 3, multinational, open-label, and randomised clinical study of patients with stage IIIB, IIIC, and IV melanoma that was not considered to be surgically resectable. Previous systemic treatment for melanoma was allowed but not required. Patients with active brain metastases, bone metastases, extensive visceral disease, primary ocular or mucosal melanoma, evidence of immunosuppression, or receiving treatment with a systemic anti-herpetic agent were excluded from the study.
Patients were randomised in a 2:1 ratio to receive either Imlygic or GM-CSF (N=436; 295 Imlygic, 141 GM-CSF). Imlygic was administered by intralesional injection at an initial concentration of 106 (1 million) PFU/mL on day 1, followed by a concentration of 108 (100 million) PFU/mL on day 21 and every 2 weeks thereafter at a dose of up to 4 mL. GM-CSF was administered subcutaneously at 125 μg/m² delivered daily for 14 days followed by a 14-day rest period in repeating intervals.
To allow for delayed immune-mediated anti-tumour effects to occur, patients were treated for a minimum of 6 months or until there were no longer any injectable lesions. During this period, treatment was to continue despite an increase in size in existing lesion(s) and/or development of new lesion(s) unless the patient developed intolerable toxicity or the investigator believed that it was in the best interest of the patient to stop treatment or to be given other therapy for melanoma. After 6 months of treatment, patients were to continue treatment until clinically relevant disease progression (i.e. disease progression associated with a decline in performance status and/or alternative therapy was required in the opinion of the investigator). Patients experiencing a response at 12 months of treatment could continue treatment for up to an additional 6 months. The mean (SD) treatment duration for the intent-to-treat (ITT) population was 15.76 weeks (15.79) in the GM-CSF arm and 26.83 weeks (18.39) in the Imlygic arm. The primary endpoint was durable response rate (DRR) [defined as the percent of patients with complete response (CR) or partial response (PR) maintained continuously for a minimum of 6 months] per blinded central review. The secondary endpoints included overall survival (OS), overall response rate (ORR) [PR+CR], time to response, duration of response, and time to treatment failure (time from randomisation until the first episode of clinically relevant disease progression where there is no response achieved after the progression event, or until death).
The mean age was 63 (range: 22 to 94) years; 26.5% were over 65 years old and 23.3% were over 74 years old. The majority of patients, 98%, were caucasian. Male patients comprised 57% of study population and 70% of patients were baseline ECOG 0 performance status. Of the enrolled patients, 22% had stage IV M1c disease and 53% of patients had received prior therapy for melanoma such as chemotherapy and cytokine-based immunotherapy in addition to surgery, adjuvant therapy or radiation. Overall, 58% of all patients enrolled into the study were seropositive for wild-type HSV-1 at baseline and 32.6% were seronegative; the HSV-1 serostatus of the remaining 9.4% was unknown.
The difference in DRR between Imlygic and GM-CSF in the ITT population was statistically significant (see table 4) in favour of Imlygic.
Table 4. Summary of results for the ITT population from Imlygic study 005/05:
| Study endpoint | Imlygic N=295 | GM-CSF N=141 | |
|---|---|---|---|
| Durable response rate | Primary | 16.3% (n=48) (95% CI: 12.1, 20.5) | 2.1% (n=3) (95% CI: 0.0, 4.5) |
| Odds ratio 8.9, (95% CI: 2.7, 29.2) P<0.0001 | |||
| Overall response rate (% CR, % PR) | Secondary | 26.4% (n=78) (95% CI: 21.4%, 31.5%) (10.8% CR, 15.6% PR) | 5.7% (n=8) (95% CI: 1.9%, 9.5%) (0.7% CR, 5% PR) |
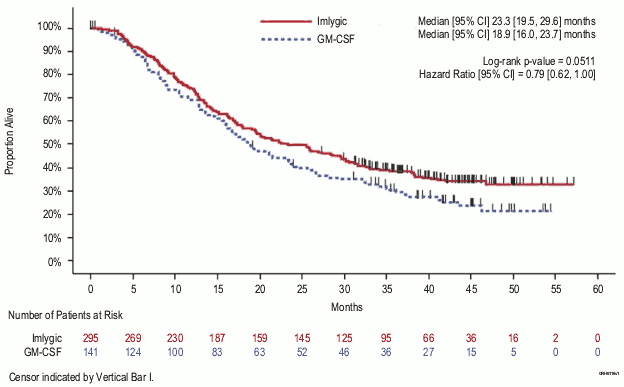
| Overall survival | Secondary | Median 23.3 (95% CI: 19.5, 29.6) months | Median 18.9 (95% CI: 16.0, 23.7) months |
| HR: 0.79, (95% CI: 0.62, 1.00) p=0.051 | |||
| Duration of response (ongoing response at last tumour evaluation) | Secondary | Not reached (Range: >0.0 έως >16.8 months) | Median 2.8 months (Range: 1.2 to >14.9 months) |
| HR: 0.46, (95% CI: 0.35, 0.60) | |||
| Time to response (median) | Secondary | 4.1 months | 3.7 months |
| Time to treatment failure (median) | Secondary | 8.2 months (95% CI: 6.5, 9.9) | 2.9 months (95% CI: 2.8, 4.0) |
| HR: 0.42, (95% CI: 0.32, 0.54) | |||
Among the Imlygic-treated responders, 56 (72%) responses were still ongoing at the time of primary analysis. Of the responders, 42 (54%) experienced a ≥25% increase in overall size of existing lesion(s) and/or developed a new lesion(s) prior to ultimately achieving a response.
In an analysis to evaluate systemic activity of Imlygic, 27 of 79 patients (34.2%) had a ≥50% overall decrease in non-visceral lesions that were not injected with Imlygic, and 8 of 71 patients (11.3%) had a ≥50% overall decrease in visceral lesions that were not injected with Imlygic.
Figure 4. Kaplan-Meier plot–overall survival (ITT population):
No overall differences in safety or efficacy were observed between elderly (≥65 years old) and younger adult patients.
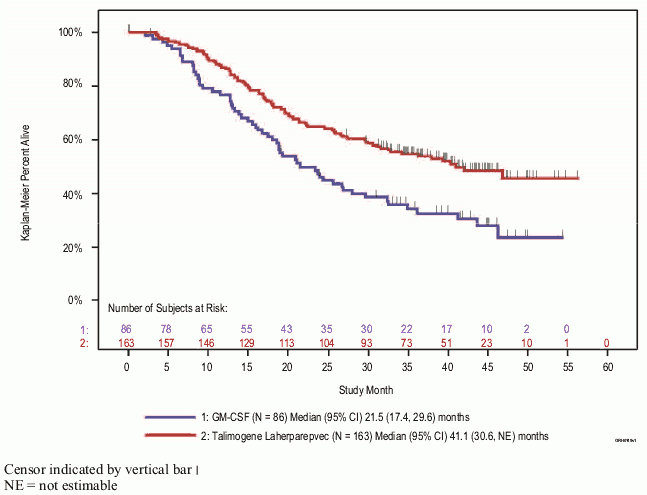
Exploratory subgroup analyses for DRR and overall survival by stage of disease were also carried out (see figure 5 and table 5). While the pivotal study was not powered to evaluate efficacy in these individual subgroups, patients with no visceral disease derived greater benefit from Imlygic treatment than those with more advanced disease.
Table 5. Summary of results from exploratory subgroup analysis from Imlygic study 005/05:
| DRR, (%) | ORR, (%) | OS (hazard ratio) | |||
|---|---|---|---|---|---|
| Imlygic | GM-CSF | Imlygic | GM-CSF | Imlygic vs GM-CSF | |
| Stage§ IIIB/IIIC/stage IVM1a (Imlygic, n=163, GM-CSF, n=86) | 25.2 | 1.2 | 40.5 | 2.3 | 0.57, (95% CI: 0.40, 0.80) |
| Stage§ IVM1B/IVM1C (Imlygic, n=131, GM-CSF, n=55) | 5.3 | 3.6 | 9.2 | 10.9 | 1.07, (95% CI: 0.75, 1.52) |
§ American Joint Committee on Cancer (AJCC) staging 6th edition.
Figure 5. Kaplan-Meier estimate of overall survival by randomised treatment arm for disease stage IIIB/IIIC/stage IVM1a (exploratory subgroup analysis):
Due to the exploratory nature of the analysis and based on the current evidence, it has not been established that Imlygic is associated with an effect on overall survival.
The European Medicines Agency has deferred the obligation to submit the results of studies with Imlygic in one or more subsets of the paediatric population in melanoma (see section 4.2 for information on paediatric use).
Talimogene laherparepvec is a genetically modified and replication-competent HSV-1 virus. Therefore, its pharmacokinetics and biodistribution are driven by the site of intralesional injection, tumour-selective replication, and release from tumour tissue.
Cellular uptake of talimogene laherparepvec occurs through HSV-1 receptors on tumours and non-tumour cells following local injection into tumours. As talimogene laherparepvec is injected and replicates intratumourally, bioavailability and systemic concentration of talimogene laherparepvec are not predictive of drug substance activity and therefore have not been evaluated.
Talimogene laherparepvec is cleared through general host-defence mechanisms (e.g. autophagy, adaptive immune responses). Talimogene laherparepvec is degraded by typical endogenous protein and DNA catabolic pathways. As with other wild-type HSV-1 infections, a latent pool of talimogene laherparepvec DNA may persist in neuronal cell bodies innervating the injection sites; therefore, the occurrence of latent infection with talimogene laherparepvec cannot be excluded.
Talimogene laherparepvec DNA was quantified with a highly sensitive and specific quantitative Polymerase Chain Reaction (qPCR) assay which may not correlate with viral infectivity risk. Talimogene laherparepvec was also quantified in selected patient samples in clinical studies using viral infectivity assays at the injection sites and in some cases of potential herpetic lesions.
The biodistribution and shedding of intralesionally administered talimogene laherparepvec were investigated in a clinical study that measured talimogene laherparepvec DNA in blood, urine, injection site, exterior of the occlusive dressings, oral mucosa, anogenital area, and suspected herpetic lesions. Sixty patients with melanoma received Imlygic intralesional injection at a dose and schedule same as clinical study 005/05 (see section 5.1). Occlusive dressings samples were collected during treatment. Blood and urine samples were collected during treatment and for up to 30 days after the end of treatment. Injection site, oral mucosa, and anogenital area samples were collected during treatment and for up to 60 days after the end of treatment. Suspected herpetic lesion samples were collected any time a patient experienced lesions of suspected herpetic origin. If the qPCR testing for talimogene laherparepvec DNA was positive, then a TCID 50 assay was performed to measure viral infectivity. In the 60 patients treated, data indicate that talimogene laherparepvec DNA was present in all sites during the study (see table 6).
Table 6. Patients with detectable DNA during treatment:
| Body fluid/site | Patients with detectable DNA during treatment (n=60) |
|---|---|
| Blood | 59 (98%) |
| Urine | 19 (32%) |
| Injection site | 60 (100%) |
| Exterior of the occlusive dressing | 48 (80%) |
| Oral mucosa | 8 (13%) |
| Anogenital area | 5 (19%)a |
a For the anogenital area, 26 patients were tested for Imlygic DNA.
The proportion of samples and subjects with talimogene laherparepvec DNA was highest during cycle 2 of treatment for the blood, urine, injection site, and occlusive dressings; highest in cycle 1 of treatment for the oral mucosa; and highest in cycles 1 and 2 for the anogenital area. Among patients with detectable talimogene laherparepvec DNA in the blood, urine, oral mucosa, and anogenital area, no samples had detectable talimogene laherparepvec DNA 30 days after the end of treatment. For patients with detectable DNA in injected lesions, no samples had detectable talimogene laherparepvec DNA 60 days after end of treatment.
Overall 3 of 19 patients with lesions of suspected herpetic origin had talimogene laherparepvec DNA present at any time during the study. Viral activity was measured in samples that were positive for talimogene laherparepvec DNA from the injection site, occlusive dressings, oral mucosa, anogenital area, and suspected herpetic lesions. No viral activity was detected in samples of the occlusive dressings, oral mucosa, anogenital area, and suspected herpetic lesions. Infectious talimogene laherparepvec virus was detected at the site of injection in 7 (11%) patients at multiple time points in the study; no samples were positive for viral infectivity after cycle 2 or after the end of treatment.
No pharmacokinetic studies using talimogene laherparepvec have been conducted in special populations.
At doses up to 4 × 108 PFU/kg or 107 PFU/dose (60-fold over the highest proposed clinical dose), single or repeated doses of talimogene laherparepvec administered by SC, IV, or intratumoural injection were well tolerated in immunocompetent mice, rats, and dogs. No neuropathology or adverse neurological effects were observed. In an in vivo study of intracerebral injection, talimogene laherparepvec was 10,000-fold less neurovirulent as compared to the wild-type HSV-1 dose that results in death 50% of the time in mice.
Talimogene laherparepvec was injected into various xenograft tumours at doses up to 2 × 108 PFU/kg (30-fold over the highest proposed clinical dose) in immunodeficient mice (nude and SCID). Lethal systemic viral infection was observed in up to 20% of nude mice (primarily deficient in T lymphocyte function) and 100% of SCID mice (devoid of both T and B lymphocytes).
Across studies, fatal disseminated viral infection was observed in 14% of nude mice following treatment with talimogene laherparepvec at doses that are 10 to 100-fold higher than those that result in 100% lethality with wild-type HSV-1.
The genotoxic potential of talimogene laherparepvec has not been evaluated in long-term animal or human studies. Because wild-type HSV-1 does not integrate into the host genome, the risk of insertional mutagenesis with talimogene laherparepvec is negligible.
The carcinogenic potential of talimogene laherparepvec has not been evaluated in long-term animal or human studies. However, available data for talimogene laherparepvec and wild-type HSV-1 do not indicate a carcinogenic risk in humans.
There were no impacts to male or female reproductive tissues following treatment of adult mice at doses up to 4 × 108 PFU/kg (60-fold higher, on a PFU/kg basis, compared to the maximum clinical dose). No effects on embryo-foetal development were observed when talimogene laherparepvec was administered during organogenesis to pregnant mice at doses up to 4 × 108 (400 million) PFU/kg (60-fold higher, on a PFU/kg basis, compared to the maximum clinical dose). Negligible amounts (<0.001% of maternal blood levels) of talimogene laherparepvec DNA were found in foetal blood.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.