Source: European Medicines Agency (EU) Revision Year: 2019 Publisher: Celgene Europe B.V., Winthontlaan 6 N, 3526 KV, Utrecht, Netherlands
For information on other medicinal products given in combination with Imnovid, refer to the respective current SmPC.
Pomalidomide must not be taken during pregnancy, since a teratogenic effect is expected. Pomalidomide is structurally related to thalidomide. Thalidomide is a known human teratogen that causes severe life-threatening birth defects. Pomalidomide was found to be teratogenic in both rats and rabbits when administered during the period of major organogenesis (see section 5.3).
The conditions of the Pregnancy Prevention Programme must be fulfilled for all patients unless there is reliable evidence that the patient does not have childbearing potential.
A female patient or a female partner of a male patient is considered of non-childbearing potential if she meets at least one of the following criteria:
For women of childbearing potential, pomalidomide is contraindicated unless all of the following are met:
The prescriber must ensure that for women of childbearing potential:
For male patients taking pomalidomide, pharmacokinetic data has demonstrated that pomalidomide is present in human semen during treatment. As a precaution, and taking into account special populations with potentially prolonged elimination time such as hepatic impairment, all male patients taking pomalidomide must meet the following conditions:
Women of childbearing potential must use at least one effective method of contraception for at least 4 weeks before therapy, during therapy, and until at least 4 weeks after pomalidomide therapy and even in case of dose interruption unless the patient commits to absolute and continuous abstinence confirmed on a monthly basis. If not established on effective contraception, the patient must be referred to an appropriately trained health care professional for contraceptive advice in order that contraception can be initiated.
The following can be considered to be examples of suitable methods of contraception:
Because of the increased risk of venous thromboembolism in patients with multiple myeloma taking pomalidomide and dexamethasone, combined oral contraceptive pills are not recommended (see also section 4.5). If a patient is currently using combined oral contraception the patient should switch to one of the effective methods listed above. The risk of venous thromboembolism continues for 4-6 weeks after discontinuing combined oral contraception. The efficacy of contraceptive steroids may be reduced during cotreatment with dexamethasone (see section 4.5).
Implants and levonorgestrel-releasing intrauterine systems are associated with an increased risk of infection at the time of insertion and irregular vaginal bleeding. Prophylactic antibiotics should be considered particularly in patients with neutropenia.
Insertion of copper-releasing intrauterine devices is not recommended due to the potential risks of infection at the time of insertion and menstrual blood loss which may compromise patients with severe neutropenia or severe thrombocytopenia.
According to local practice, medically supervised pregnancy tests with a minimum sensitivity of 25 mIU/mL must be performed for women of childbearing potential as outlined below. This requirement includes women of childbearing potential who practice absolute and continuous abstinence. Ideally, pregnancy testing, issuing a prescription and dispensing should occur on the same day. Dispensing of pomalidomide to women of childbearing potential should occur within 7 days of the prescription.
A medically supervised pregnancy test should be performed during the consultation, when pomalidomide is prescribed, or in the 3 days prior to the visit to the prescriber once the patient had been using effective contraception for at least 4 weeks. The test should ensure the patient is not pregnant when she starts treatment with pomalidomide.
A medically supervised pregnancy test should be repeated at least every 4 weeks, including at least 4 weeks after the end of treatment, except in the case of confirmed tubal sterilisation. These pregnancy tests should be performed on the day of the prescribing visit or in the 3 days prior to the visit to the prescriber.
Patients should be instructed never to give this medicinal product to another person and to return any unused capsules to their pharmacist at the end of treatment.
Patients should not donate blood, semen or sperm during treatment (including during dose interruptions) and for 7 days following discontinuation of pomalidomide.
In order to assist patients in avoiding foetal exposure to pomalidomide, the Marketing Authorisation Holder will provide educational material to health care professionals to reinforce the warnings about the expected teratogenicity of pomalidomide, to provide advice on contraception before therapy is started, and to provide guidance on the need for pregnancy testing. The prescriber must inform the patient about the expected teratogenic risk and the strict pregnancy prevention measures as specified in the Pregnancy Prevention Programme and provide patients with appropriate patient educational brochure, patient card and/or equivalent tool in accordance with the national implemented patient card system. A national controlled distribution system has been implemented in collaboration with each National Competent Authority. The controlled distribution system includes the use of a patient card and/or equivalent tool for prescribing and /or dispensing controls, and the collection of detailed data relating to the indication in order to monitor the off-label use within the national territory. Ideally, pregnancy testing, issuing a prescription and dispensing should occur on the same day. Dispensing of pomalidomide to women of childbearing potential should occur within 7 days of the prescription and following a medically supervised negative pregnancy test result. Prescriptions for women of childbearing potential can be for a maximum duration of 4 weeks, and prescriptions for all other patients can be for a maximum duration of 12 weeks.
Neutropenia was the most frequently reported Grade 3 or 4 haematological adverse reaction in patients with relapsed/refractory multiple myeloma, followed by anaemia and thrombocytopenia. Patients should be monitored for haematological adverse reactions, especially neutropenia. Patients should be advised to report febrile episodes promptly. Physicians should observe patients for signs of bleeding including epistaxes, especially with use of concomitant medicinal products known to increase the risk of bleeding (see section 4.8). Complete blood counts should be monitored at baseline, weekly for the first 8 weeks and monthly thereafter. A dose modification may be required (see section 4.2). Patients may require use of blood product support and/or growth factors.
Patients receiving pomalidomide either in combination with bortezomib and dexamethasone or in combination with dexamethasone have developed venous thromboembolic events (predominantly deep vein thrombosis and pulmonary embolism) and arterial thrombotic events (myocardial infarction and cerebrovascular accident). Patients with known risk factors for thromboembolism – including prior thrombosis – should be closely monitored. Action should be taken to try to minimise all modifiable risk factors (e.g. smoking, hypertension, and hyperlipidaemia). Patients and physicians are advised to be observant for the signs and symptoms of thromboembolism. Patients should be instructed to seek medical care if they develop symptoms such as shortness of breath, chest pain, arm or leg swelling. Anticoagulation therapy (unless contraindicated) is recommended, (such as acetylsalicylic acid, warfarin, heparin or clopidogrel), especially in patients with additional thrombotic risk factors. A decision to take prophylactic measures should be made after a careful assessment of the individual patient’s underlying risk factors. In clinical studies, patients received prophylactic acetylsalicylic acid or alternative antithrombotic therapy. The use of erythropoietic agents carries a risk of thrombotic events including thromboembolism. Therefore, erythropoietic agents, as well as other agents that may increase the risk of thromboembolic events, should be used with caution.
Patients with ongoing ≥ Grade 2 peripheral neuropathy were excluded from clinical studies with pomalidomide. Appropriate caution should be exercised when considering the treatment of such patients with pomalidomide.
Patients with significant cardiac dysfunction (congestive heart failure [NY Heart Association Class III or IV]; myocardial infarction within 12 months of starting study; unstable or poorly controlled angina pectoris) were excluded from clinical studies with pomalidomide. Cardiac events, including congestive cardiac failure, pulmonary oedema and atrial fibrillation (see section 4.8), have been reported, mainly in patients with pre-existing cardiac disease or cardiac risk factors. Appropriate caution should be exercised when considering the treatment of such patients with pomalidomide, including periodic monitoring for signs or symptoms of cardiac events.
Patients at greatest risk of tumour lysis syndrome are those with high tumour burden prior to treatment. These patients should be monitored closely and appropriate precautions taken.
Second primary malignancies, such as non-melanoma skin cancer, have been reported in patients receiving pomalidomide (see section 4.8). Physicians should carefully evaluate patients before and during treatment using standard cancer screening for occurrence of second primary malignancies and institute treatment as indicated.
Angioedema and severe dermatologic reactions including SJS, TEN and DRESS have been reported with the use of pomalidomide (see section 4.8). Patients should be advised of the signs and symptoms of these reactions by their prescribers and should be told to seek medical attention immediately if they develop these symptoms. Pomalidomide must be discontinued for exfoliative or bullous rash, or if SJS, TEN or DRESS is suspected, and should not be resumed following discontinuation for these reactions. Patients with a prior history of serious allergic reactions associated with thalidomide or lenalidomide were excluded from clinical studies. Such patients may be at higher risk of hypersensitivity reactions and should not receive pomalidomide. Pomalidomide interruption or discontinuation should be considered for Grade 2-3 skin rash. Pomalidomide must be discontinued permanently for angioedema.
Dizziness and confusional state have been reported with pomalidomide. Patients must avoid situations where dizziness or confusion may be a problem and not to take other medicinal products that may cause dizziness or confusion without first seeking medical advice.
ILD and related events, including cases of pneumonitis, have been observed with pomalidomide. Careful assessment of patients with an acute onset or unexplained worsening of pulmonary symptoms should be performed to exclude ILD. Pomalidomide should be interrupted pending investigation of these symptoms and if ILD is confirmed, appropriate treatment should be initiated. Pomalidomide should only be resumed after a thorough evaluation of the benefits and the risks.
Markedly elevated levels of alanine aminotransferase and bilirubin have been observed in patients treated with pomalidomide (see section 4.8). There have also been cases of hepatitis that resulted in discontinuation of pomalidomide. Regular monitoring of liver function is recommended for the first 6 months of treatment with pomalidomide and as clinically indicated thereafter.
Reactivation of hepatitis B has been reported rarely in patients receiving pomalidomide in combination with dexamethasone who have previously been infected with the hepatitis B virus (HBV). Some of these cases have progressed to acute hepatic failure, resulting in discontinuation of pomalidomide. Hepatitis B virus status should be established before initiating treatment with pomalidomide. For patients who test positive for HBV infection, consultation with a physician with expertise in the treatment of hepatitis B is recommended. Caution should be exercised when pomalidomide in combination with dexamethasone is used in patients previously infected with HBV, including patients who are anti-HBc positive but HBsAg negative. These patients should be closely monitored for signs and symptoms of active HBV infection throughout therapy.
This medicinal product contains less than 1 mmol sodium (23 mg) per capsule, i.e. essentially ‘sodium-free’.
For information on other medicinal products given in combination with Imnovid, refer to the respective current SmPC.
Pomalidomide is not anticipated to cause clinically relevant pharmacokinetic drug-drug interactions due to P450 isoenzyme inhibition or induction or transporter inhibition when co-administered with substrates of these enzymes or transporters. The potential for such drug-drug interactions, including the potential impact of pomalidomide on the pharmacokinetics of combined oral contraceptives, has not been evaluated clinically (see section 4.4 Teratogenicity).
Pomalidomide is partly metabolised by CYP1A2 and CYP3A4/5. It is also a substrate for P-glycoprotein. Co-administration of pomalidomide with the strong CYP3A4/5 and P-gp inhibitor ketoconazole, or the strong CYP3A4/5 inducer carbamazepine, had no clinically relevant effect on exposure to pomalidomide. Co-administration of the strong CYP1A2 inhibitor fluvoxamine with pomalidomide in the presence of ketoconazole, increased mean exposure to pomalidomide by 107% with a 90% confidence interval [91% to 124%] compared to pomalidomide plus ketoconazole. In a second study to evaluate the contribution of a CYP1A2 inhibitor alone to metabolism changes, co-administration of fluvoxamine alone with pomalidomide increased mean exposure to pomalidomide by 125% with a 90% confidence interval [98% to 157%] compared to pomalidomide alone. If strong inhibitors of CYP1A2 (e.g. ciprofloxacin, enoxacin and fluvoxamine) are co-administered with pomalidomide, reduce the dose of pomalidomide by 50%.
Co-administration of multiple doses of up to 4 mg pomalidomide with 20 mg to 40 mg dexamethasone (a weak to moderate inducer of several CYP enzymes including CYP3A) to patients with multiple myeloma had no effect on the pharmacokinetics of pomalidomide compared with pomalidomide administered alone.
The effect of dexamethasone on warfarin is unknown. Close monitoring of warfarin concentration is advised during treatment.
For information on other medicinal products given in combination with Imnovid, refer to the respective current SmPC.
Women of childbearing potential should use effective method of contraception. If pregnancy occurs in a woman treated with pomalidomide, treatment must be stopped and the patient should be referred to a physician specialised or experienced in teratology for evaluation and advice. If pregnancy occurs in a partner of a male patient taking pomalidomide, it is recommended to refer the female partner to a physician specialised or experienced in teratology for evaluation and advice. Pomalidomide is present in human semen. As a precaution, all male patients taking pomalidomide should use condoms throughout treatment duration, during dose interruption and for 7 days after cessation of treatment if their partner is pregnant or of childbearing potential and has no contraception (see sections 4.3 and 4.4).
A teratogenic effect of pomalidomide in humans is expected. Pomalidomide is contraindicated during pregnancy and in women of childbearing potential, except when all the conditions for pregnancy prevention have been met, see section 4.3 and section 4.4.
It is unknown whether pomalidomide is excreted in human milk. Pomalidomide was detected in milk of lactating rats following administration to the mother. Because of the potential for adverse reactions in breastfed infants from pomalidomide, a decision must be made whether to discontinue breast-feeding or to discontinue the medicinal product, taking into account the benefit of breast-feeding for the child and the benefit of the therapy for the woman.
Pomalidomide was found to impact negatively on fertility and be teratogenic in animals. Pomalidomide crossed the placenta and was detected in foetal blood following administration to pregnant rabbits, see section 5.3.
Pomalidomide has minor or moderate influence on the ability to drive and use machines. Fatigue, depressed level of consciousness, confusion, and dizziness have been reported with the use of pomalidomide. If affected, patients should be instructed not to drive cars, use machines or perform hazardous tasks while being treated with pomalidomide.
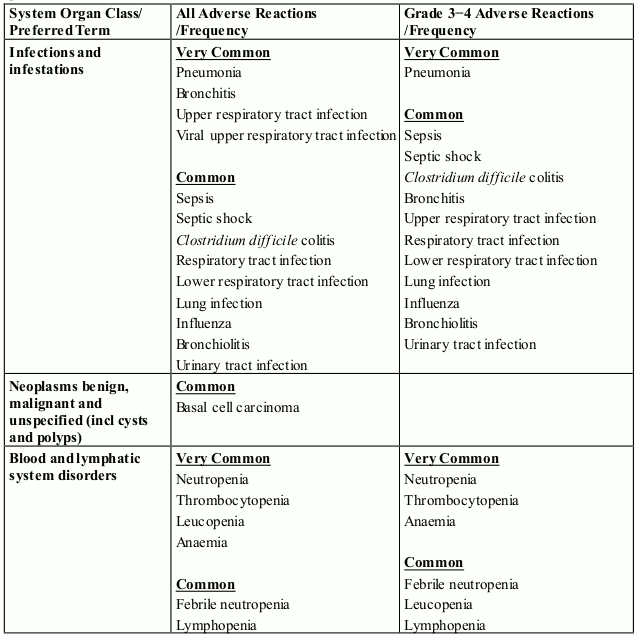
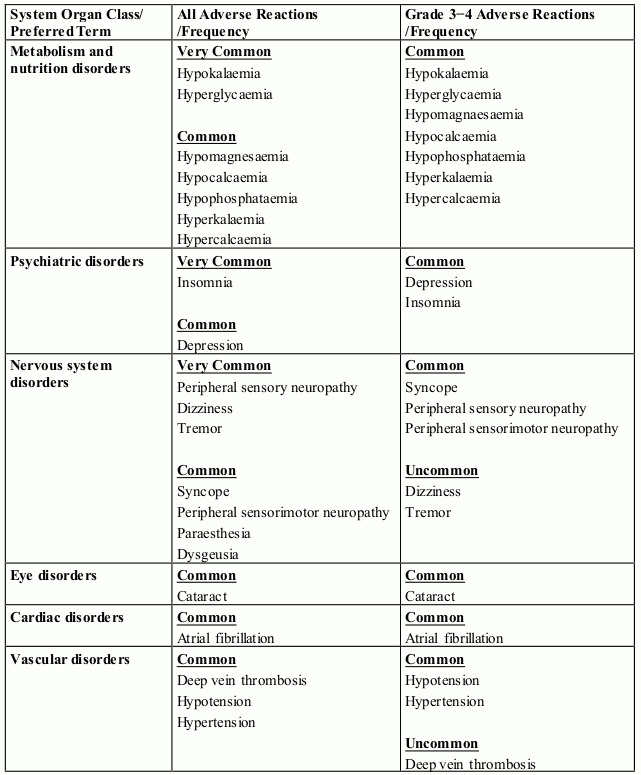
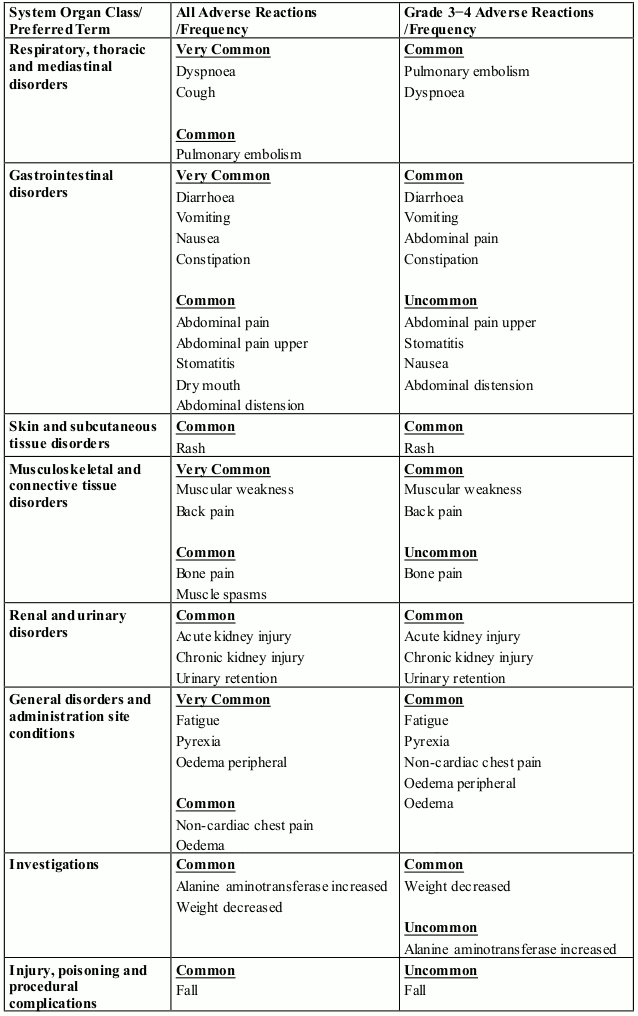
The most commonly reported blood and lymphatic system disorders were neutropenia (46.8%), thrombocytopenia (36.7%) and anaemia (28.4%). The most frequently reported adverse reaction was peripheral sensory neuropathy (47.8%). The most commonly reported Grade 3 or 4 adverse reactions were blood and lymphatic system disorders including neutropenia (41.7%), thrombocytopenia (27.3%) and anaemia (14.0%). The most commonly reported serious adverse reaction was pneumonia (11.5%). Other serious adverse reactions reported included pyrexia (4.0%), lower respiratory tract infection (2.9%), pulmonary embolism (2.9%), influenza (2.9%), and acute kidney injury (2.9%).
The most commonly reported adverse reactions in clinical studies have been blood and lymphatic system disorders including anaemia (45.7%), neutropenia (45.3%) and thrombocytopenia (27%); in general disorders and administration site conditions including fatigue (28.3%), pyrexia (21%) and oedema peripheral (13%); and in infections and infestations including pneumonia (10.7%). Peripheral neuropathy adverse reactions were reported in 12.3% of patients and venous embolic or thrombotic (VTE) adverse reactions were reported in 3.3% of patients. The most commonly reported Grade 3 or 4 adverse reactions were in the blood and lymphatic system disorders including neutropenia (41.7%), anaemia (27%) and thrombocytopenia (20.7%); in infections and infestations including pneumonia (9%); and in general disorders and administration site conditions including fatigue (4.7%), pyrexia (3%) and oedema peripheral (1.3%). The most commonly reported serious adverse reaction was pneumonia (9.3%). Other serious adverse reactions reported included febrile neutropenia (4.0%), neutropenia (2.0%), thrombocytopenia (1.7%) and VTE adverse reactions (1.7%).
Adverse reactions tended to occur more frequently within the first 2 cycles of treatment with pomalidomide.
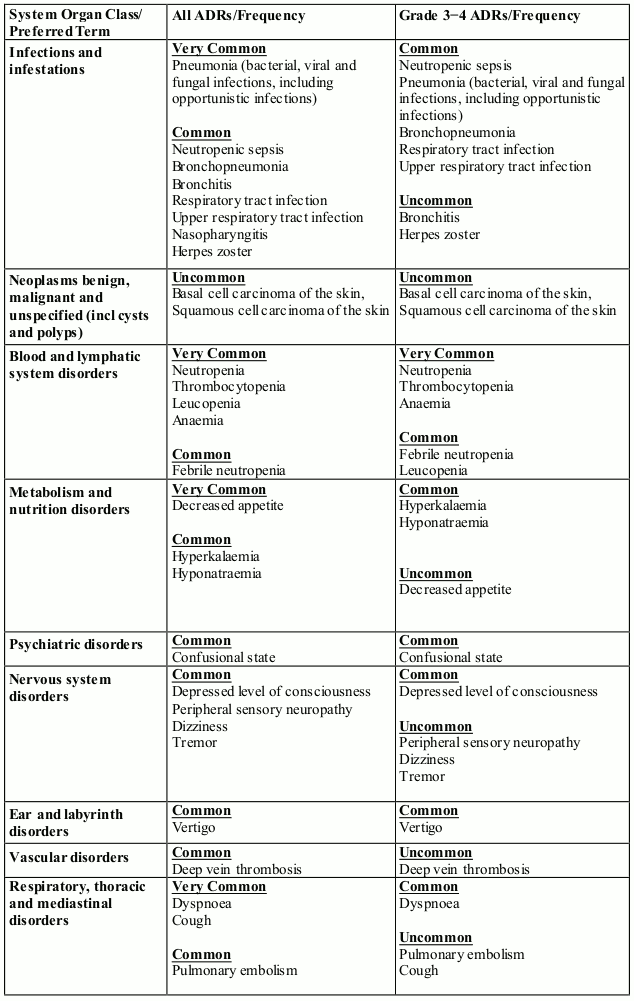
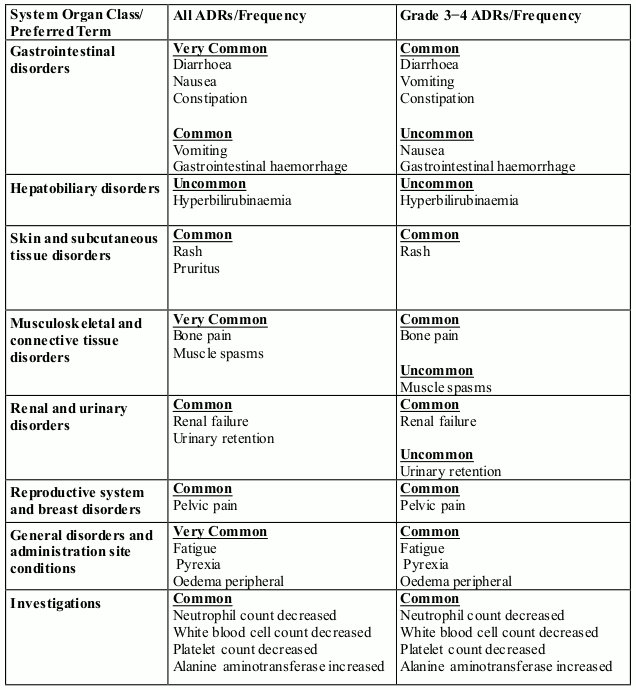
In randomised study CC-4047-MM-007, 278 patients received pomalidomide, bortezomib and dexamethasone (Pom+Btz+Dex arm). See section 4.2 for dosing information.
The adverse reactions observed in patients treated with pomalidomide in combination with bortezomib and dexamethasone are listed in Table 7 by system organ class (SOC) and frequency for all adverse reactions and for Grade 3 or 4 adverse reactions.
Frequencies for Pom+Btz+Dex (any grade) are defined in accordance with current guidance, as: very common (≥1/10), common (≥1/100 to <1/10); and uncommon (≥1/1,000 to <1/100).
Table 7. All Adverse Reactions (ADRs) reported in clinical trial MM-007 in patients treated with pomalidomide in combination with bortezomib and dexamethasone:
In randomised study CC-4047-MM-003, 302 patients with relapsed and refractory multiple myeloma were exposed to 4 mg pomalidomide administered once daily for 21 days of each 28–day cycle in combination with a weekly low dose of dexamethasone.
The adverse reactions observed in patients treated with pomalidomide plus dexamethasone are listed below in Table 8 by system organ class (SOC) and frequency for all adverse reactions (ADRs) and for Grade 3 or 4 adverse reactions.
The frequencies of adverse reactions are those reported in the pomalidomide plus dexamethasone arm of study CC-4047-MM-003 (n=302). Within each SOC and frequency grouping, adverse reactions are presented in order of decreasing seriousness. Frequencies are defined in accordance with current guidance, as: very common (≥1/10), common (≥1/100 to <1/10); and uncommon (≥1/1,000 to <1/100).
Table 8. ADRs reported in clinical study MM-003 in patients treated with pomalidomide in combination with dexamethasone:
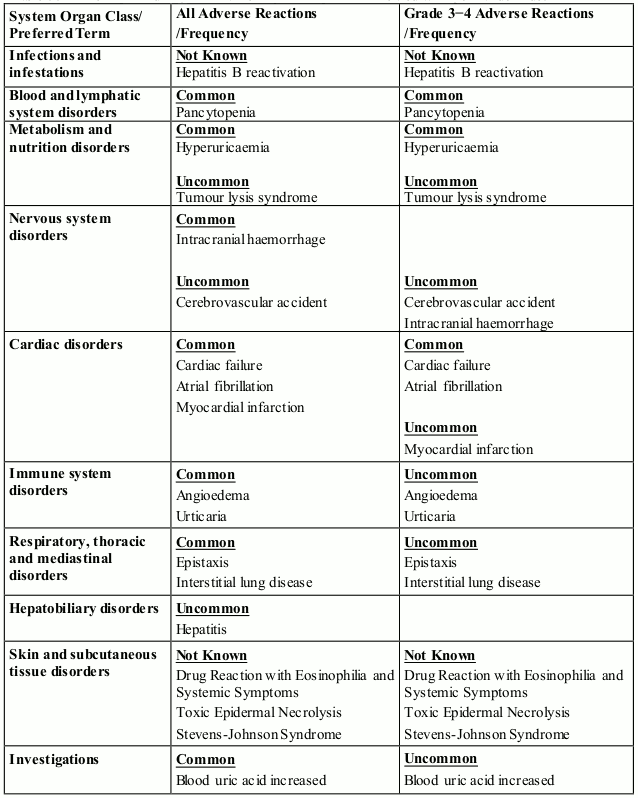
In addition to the above adverse reactions identified from the pivotal clinical trials, the following Table 9 is derived from data gathered from post-marketing surveillance.
Table 9. ADRs reported in post-marketing use in patients treated with pomalidomide:
Pomalidomide is structurally related to thalidomide. Thalidomide is a known human teratogenic active substance that causes severe life-threatening birth defects. Pomalidomide was found to be teratogenic in both rats and rabbits when administered during the period of major organogenesis (see sections 4.6 and 5.3). If pomalidomide is taken during pregnancy, a teratogenic effect of pomalidomide in humans is expected (see section 4.4).
In patients receiving combination therapy with pomalidomide in clinical studies, neutropenia occurred in up to 46.8% of patients (41.7% Grade 3 or 4). Neutropenia did not lead to pomalidomide discontinuation in any patient and was infrequently serious.
Febrile neutropenia (FN) was reported in 3.2-6.7% of patients and was serious in 1.8-4.0% of patients (see section 4.2 and 4.4).
In patients receiving combination therapy with pomalidomide in clinical studies, thrombocytopenia occurred in 27.0-36.7% of patients. Thrombocytopenia was Grade 3 or 4 in 20.7-27.3% of patients, led to pomalidomide discontinuation in 0.7% of patients and was serious in 0.4-1.7% of patients (see sections 4.2 and 4.4).
Neutropenia and thrombocytopenia tended to occur more frequently within the first 2 cycles of treatment with pomalidomide.
Infection was the most common non haematological toxicity.
In patients receiving combination therapy with pomalidomide in clinical studies, infection occurred in 55.0-80.2% of patients (24.0-30.9% Grade 3 or 4). Upper respiratory tract infection and pneumonia were the most frequently occurring infections. Fatal infections (Grade 5) occurred in 2.7-4.0% of patients. Infections led to pomalidomide discontinuation in 2.0-2.9% of patients.
Prophylaxis with acetylsalicylic acid (and other anticoagulants in high risk patients) was mandatory for all patients in clinical studies. Anticoagulation therapy (unless contraindicated) is recommended (see section 4.4).
In patients receiving combination therapy with pomalidomide in clinical studies, venous thromboembolic events (VTE) occurred in 3.3-11.5% of patients (1.3-5.4% Grade 3 or 4). VTE was reported as serious in 1.7-4.3% of patients, no fatal reactions were reported, and VTE was associated with pomalidomide discontinuation in up to 1.8% of patients.
Patients with ongoing peripheral neuropathy ≥ Grade 2 with pain within 14 days prior to randomisation were excluded from clinical trials. Peripheral neuropathy occurred in 55.4 % of patients (10.8% Grade 3; 0.7% Grade 4). Exposure-adjusted rates were comparable across treatment arms. Approximately 30% of the patients experiencing peripheral neuropathy had a history of neuropathy at baseline. Peripheral neuropathy led to discontinuation of bortezomib in approximately 12.9% of patients, pomalidomide in 1.8% and dexamethasone in 2.2-8.9% of patients, respectively. Refer also to the bortezomib SmPC.
Patients with ongoing peripheral neuropathy ≥ Grade 2 were excluded from clinical studies. Peripheral neuropathy occurred in 12.3% of patients (1.0% Grade 3 or 4). No peripheral neuropathy reactions were reported as serious, and peripheral neuropathy led to dose discontinuation in 0.3% of patients (see section 4.4).
Haemorrhagic disorders have been reported with pomalidomide, especially in patients with risk factors such as concomitant medicinal products that increase susceptibility to bleeding. Haemorrhagic events have included epistaxis, intracranial haemorrhage and gastrointestinal haemorrhage.
Angioedema and severe cutaneous reactions including SJS, TEN and DRESS has been reported with the use of pomalidomide. Patients with a history of severe rash associated with lenalidomide or thalidomide should not receive pomalidomide (see section 4.4).
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed in Appendix V.
Not applicable.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.