INLURIYO Film-coated tablet Ref.[116183] Active ingredients: Imlunestrant
Source: European Medicines Agency (EU) Revision Year: 2026 Publisher: Eli Lilly Nederland B.V., Papendorpseweg 83, 3528 BJ Utrecht, The Netherlands
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Not yet assigned
ATC code: Not yet assigned
Mechanism of action
Imlunestrant is an antagonist and degrader of wild-type and mutant oestrogen receptor-α (ERα), leading to inhibition of oestrogen receptor-dependent gene transcription and cellular proliferation in ER-positive breast cancer cells.
Pharmacodynamic effects
Cardiac electrophysiology
The effect of imlunestrant monotherapy on the QTc interval was evaluated in 79 patients with matching pharmacokinetics and QTcF samples from EMBER. Results showed no effect of imlunestrant concentrations across the 200 mg to 1 200 mg dose range on QTc interval, with the upper bound of the 90% CI of the mean delta QTc less than 10 ms at Cmax of 400 mg (change from baseline of 1.72 ms; 90% CI: -0.43, 3.87).
Clinical efficacy and safety
The efficacy and safety of imlunestrant was evaluated in EMBER 3, a Phase 3 global, randomized, open-label study for adult patients with ER-positive, HER2-negative, locally advanced (not amenable to curative treatment by surgery) or metastatic breast cancer (mBC), who have been treated with an aromatase inhibitor (AI), alone or in combination with a CDK4/6 inhibitor.
Eligible patients were pre-, peri- and postmenopausal women, or men, (≥18 years of age) with ER-positive, HER2-negative advanced breast cancer that had previously received an AI alone or combined with a CDK4/6 inhibitor in the adjuvant or metastatic setting. Patients had either: experienced recurrence while receiving or within 12 months of completing adjuvant treatment with an AI alone or with a CDK4/6 inhibitor for early breast cancer, experienced recurrence >12 months after completing adjuvant treatment followed by disease progression on or after an AI alone or with a CDK4/6 inhibitor, or been diagnosed with de novo metastatic disease and had disease progression on or after an AI alone or with a CDK4/6 inhibitor. All patients were required to have Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, adequate organ function, and evaluable lesions per Response Evaluation Criteria in Solid Tumours (RECIST) version 1.1, i.e., measurable disease or bone only disease with evaluable lesions. LHRH agonists were given to pre- and perimenopausal women, and men. Patients with presence of symptomatic metastatic visceral disease, and patients with cardiac comorbidity were excluded.
Patients were enrolled regardless of ESR1-mutation status. ESR1-mutational status was determined by blood circulating tumour deoxyribonucleic acid (ctDNA) using the Guardant360 CDx assay. A result was considered ESR1-positive if at least one of 34 predefined ESR1 variants was detected: E380A, E380D, E380K, E380Q, E380V, M421_V422delinsl, V422_E423del, V422del, S463F, S463P, L469V, L536F, L536G, L536H, L536I, L536K, L536N, L536P, L536Q, L536R, L536V, Y537C, Y537D, Y537G, Y537H, Y537N, Y537P, Y537Q, Y537S, D538E, D538G, D538H, D538N, D538V. Among these, 17 variants were identified within the EMBER-3 study population: E380K, E380Q, V422del, S463P, L469V, L536H, L536K, L536P, L536Q, L536R, Y537C, Y537D, Y537N, Y537S, D538E, D538G, D538N.
A total of 874 patients were randomised 1:1:1 between 3 treatment arms: 400 mg oral daily administration of Inluriyo (Arm A), investigators choice of standard of care (SOC) (fulvestrant or exemestane) (Arm B), or 400 mg oral daily administration of Inluriyo in combination with abemaciclib (Arm C). Among the 330 patients randomised to investigators choice endocrine therapy (Arm B), 292 received fulvestrant (90%), and 32 received exemestane (10%). Randomization was stratified by previous treatment with CDK4/6 inhibitor (yes vs. no), presence of visceral metastasis (yes vs. no), and region (East Asia vs. North America/Western Europe vs. Others). The demographics and baseline disease characteristics were well balanced between treatment arms. Baseline demographics of the overall study population were as follows: median age was 61 years (range: 27-89) and 13% were ≥75 years, 99% were female, 56% were white, 30% Asian, 3% Black, and 11% were other or missing. The majority of patients were treated in the second-line advanced setting (67%) versus the first-line setting (33%), and the majority had received a prior CDK4/6i (60%), 37% received palbociclib, 15% ribociclib and 3% abemaciclib. Baseline ECOG performance was 0 (65%) or 1 (35%). Patient demographics for those with ESR1-mutated tumours were generally representative of the broader study population.
The primary efficacy endpoint was progression-free survival (PFS) as assessed by investigator. Key secondary endpoint for EMBER-3 was overall survival (OS).
EMBER 3: Inluriyo monotherapy for patients with ESR1m
At the primary analysis (24 June 2024 cut-off), a statistically significant improvement in PFS was observed in patients who received Inluriyo monotherapy versus SOC in the ESR1m subpopulation. The comparison of Inluriyo monotherapy versus SOC was not statistically significant in the ITT population. Efficacy results in the ESR1m subpopulation are provided in Table 4 and Figure 1.
Table 4. Summary of efficacy data for patients with ESR1m treated with Inluriyo monotherapy in EMBER-3:
| Inluriyo N=138 | Standard of care N=118 | |
| Progression-free survival | ||
| Number of events, n (%) | 109 (79.0) | 102 (86.4) |
| Median PFS, months (95% CI)* | 5.5 (3.9, 7.4) | 3.8 (3.7, 5.5) |
| Hazard ratio (95% CI)** | 0.617 (0.464, 0.821) | |
| p-value (2-sided)** | 0.0008 | |
CI = confidence interval; ESR1 = oestrogen receptor 1.
* Kaplan-Meier estimate; 95 % CI based on the Brookmeyer-Crowley method.
** From a Cox proportional hazards model and a stratified log-rank test stratified by previous treatment with CDK4/6 inhibitor (yes vs. no) and presence of visceral metastasis (yes vs. no).
Data cut-off date 24 June 2024
Figure 1. Kaplan-Meier Curve of progression free survival for patients with ESR1m treated with Inluriyo monotherapy in EMBER-3:
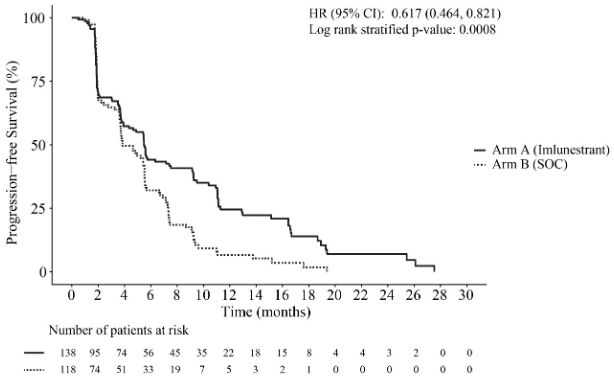
In the CDK4/6i naïve subgroup, the median PFS in the Inluriyo arm was 11.1 months (95% CI: 5.5, 16.5) compared to 5.7 months (95% CI: 3.8, 7.4) in SOC arm (HR = 0.42; 95% CI: 0.25, 0.72). In the CDK4/6i pre-treated subgroup, the median PFS in the Inluriyo arm was 3.9 months (95% CI: 2.0, 6.0) compared to 3.7 months (95% CI: 2.2, 4.6) in SOC arm (HR = 0.72; 95% CI: 0.52, 1.0).
At the third OS interim analysis (18 August 2025 cut-off), 128 events were observed across the two arms, and the HR was 0.60 (95% CI: 0.43, 0.86).
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with imlunestrant in all subsets of the paediatric population in breast cancer (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
In a population pharmacokinetic analysis, the steady-state mean (CV%) maximum concentration (Cmax) of imlunestrant is 141 ng/mL (45%) and the AUC0-24h is 2 400 ng*h/mL (46%) after administration of the recommended dose of 400 mg once daily. The Cmax and AUC of imlunestrant increase proportionally over a dose range from 200 mg to 1 200 mg once daily (0.5 to 3 times the approved recommended dose).
Absorption
The mean (CV%) absolute bioavailability of imlunestrant after a single oral 400 mg dose is 10.5% (32%). The median time to reach peak plasma concentration (tmax) is approximately 4 hours.
Effect of food
Administration of imlunestrant 400 mg with a low-fat meal (approximately 400-500 calories with 100-125 calories from fat) increased Cmax by 3.55-fold and AUC(0-∞) by 2.04-fold compared to fasted administration. Imlunestrant exposures in the presence of a high-fat meal are unknown. Distribution In a population pharmacokinetic analysis, the mean (CV%) apparent central volume of distribution of imlunestrant is 4 310 L (69%) in patients with advanced or metastatic breast cancer. Human protein binding of imlunestrant is 99.93% to 99.96% at clinically relevant concentrations.
Biotransformation
Imlunestrant is metabolized by sulfation, CYP3A4 oxidation and direct glucuronidation.
In vitro studies indicated that imlunestrant is a substrate of P-gp but not a substrate of BCRP, OCT1, OATP1B1, or OATP1B3.
Coadministration of imlunestrant with quinidine (P-gp inhibitor) had no clinically meaningful effect on the pharmacokinetics of imlunestrant.
Elimination
The elimination half-life of imlunestrant is approximately 30 hours and the mean (CV%) apparent clearance is 166 L/h (51%). Following a single radiolabelled dose of imlunestrant 400 mg to healthy subjects, 97.3% of the dose was recovered in feces and 0.278% in urine.
Special populations
Effect of age, race, and body weight
In a population pharmacokinetic analysis, age (range: 28 years to 95 years), race and body weight (range: 36 kg to 145 kg) had no clinically meaningful effect on the pharmacokinetics of imlunestrant.
Hepatic impairment
There were no clinically relevant differences in the pharmacokinetics of imlunestrant in subjects with mild hepatic impairment (Child-Pugh A). The AUC of unbound imlunestrant increased 1.82-fold in subjects with moderate hepatic impairment (Child-Pugh B) and 2.33-fold in subjects with severe hepatic impairment (Child-Pugh C).
Renal impairment
In a population pharmacokinetic analysis, mild renal impairment (60 mL/min ≤ eGFR < 90 mL/min) and moderate renal impairment (30 mL/min ≤ eGFR < 60 mL/min) had no effect on the exposure of imlunestrant. The effect of severe renal impairment (15 mL/min ≤ eGFR < 30 mL/min) suggested exposure could be increased based on limited data in 2 trial participants. Pharmacokinetics of imlunestrant in patients on dialysis is unknown.
5.3. Preclinical safety data
Repeat-dose toxicity
Repeat-dose studies were conducted in rats and non-human primates to characterize toxicity. Adverse reactions observed at clinically relevant exposure levels were ovarian cysts and marked increases in ovarian weights, atrophy of the endometrium and myometrium of the uterus, epithelium and stroma of the cervix, and epithelium of the vagina in non-human primates. Minor, non-adverse vacuolation of macrophages in the mesenteric lymph node and ileum of non-human primates occurred at exposures that were 0.8-times and 11-times greater than human exposure (AUC0-24) at 400 mg. Similar effects were observed in rats at exposures 4-times greater than human exposure (AUC0-24) at 400 mg. Other important effects in rats were hyperplasia of the urinary bladder transitional epithelium, minimal to mild renal tubular degeneration, minimal inflammation of the renal pelvis, minimal to mild ocular lens
fibre degeneration, non-adverse hypertrophy of the pituitary pars distalis in females, non-adverse decrease in pituitary gland weights, and non-adverse atrophy or hypertrophy of the adrenal cortex, which occurred at exposures at least 4-times greater than human exposure (AUC0-24) at 400 mg.
Genotoxicity
Non-clinical data reveal no special hazard for humans based on conventional studies of genotoxicity potential.
Reproductive and developmental toxicity
Based on findings in animals and its mechanism of action, imlunestrant may cause effects on male and female fertility which were generally reversible, and fetal harm when administered during pregnancy. Ovarian cysts and atrophy of the vagina, cervix, or uterus were observed in rats at and monkeys, at exposures that were 4-fold and 0.8-fold higher, respectively, than human exposure (AUC0-24) at 400 mg. Additional reproductive findings occurred in rats and consisted of cessation of estrous cycling, lobular hyperplasia or hypertrophy of the female mammary gland epithelium, spermatid retention, and cellular debris in the lumen of the epididymis at exposures (AUC0-24) at least 4-fold above that in humans. Administration of imlunestrant to pregnant rats during the period of organogenesis led to maternal toxicity, early delivery, embryo lethality, and teratogenic fetal effects at maternal exposures less than or equal to human therapeutic exposure.
Carcinogenicity
In the 26-week transgenic mouse carcinogenicity study, rasH2 mice were given oral doses of 5, 375, or 750 mg/kg imlunestrant, which caused an increased incidence of benign and malignant sex cord stromal tumours (granulosa and mixed cell) in the ovaries of mice at all dose levels. These doses correspond to 2-, 32-, and 41-times the human AUC at the recommended dose. Induction of such tumours is consistent with the pharmacology-related endocrine feedback alterations in gonadotropin levels caused by an anti-oestrogen.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.