ISENTRESS Granules for oral suspension Ref.[8807] Active ingredients: Raltegravir
Source: European Medicines Agency (EU) Revision Year: 2019 Publisher: Merck Sharp & Dohme B.V., Waarderweg 39, 2031 BN Haarlem, The Netherlands
Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in section 6.1.
Special warnings and precautions for use
General
Patients should be advised that current anti-retroviral therapy does not cure HIV and has not been proven to prevent the transmission of HIV to others through blood contact. While effective viral suppression with antiretroviral therapy has been proven to substantially reduce the risk of sexual transmission, a residual risk cannot be excluded. Precautions to prevent transmission should be taken in accordance with national guidelines.
Raltegravir has a relatively low genetic barrier to resistance. Therefore, whenever possible, raltegravir should be administered with two other active ARTs to minimise the potential for virological failure and the development of resistance (see section 5.1).
In treatment-naïve patients, the clinical study data on use of raltegravir are limited to use in combination with two nucleotide reverse transcriptase inhibitors (NRTIs) (emtricitabine and tenofovir disoproxil fumarate).
Depression
Depression, including suicidal ideation and behaviours, has been reported, particularly in patients with a pre-existing history of depression or psychiatric illness. Caution should be used in patients with a pre-existing history of depression or psychiatric illness.
Hepatic impairment
The safety and efficacy of raltegravir have not been established in patients with severe underlying liver disorders. Therefore, raltegravir should be used with caution in patients with severe hepatic impairment (see sections 4.2 and 5.2).
Patients with pre-existing liver dysfunction including chronic hepatitis have an increased frequency of liver function abnormalities during combination anti-retroviral therapy and should be monitored according to standard practice. If there is evidence of worsening liver disease in such patients, interruption or discontinuation of treatment should be considered.
Patients with chronic hepatitis B or C and treated with combination anti-retroviral therapy are at an increased risk for severe and potentially fatal hepatic adverse reactions.
Osteonecrosis
Although the aetiology is considered to be multifactorial (including corticosteroid use, alcohol consumption, severe immunosuppression, higher body mass index), cases of osteonecrosis have been reported particularly in patients with advanced HIV disease and/or long-term exposure to combination anti-retroviral therapy. Patients should be advised to seek medical advice if they experience joint aches and pain, joint stiffness or difficulty in movement.
Immune reactivation syndrome
In HIV-infected patients with severe immune deficiency at the time of institution of combination antiretroviral therapy (CART), an inflammatory reaction to asymptomatic or residual opportunistic pathogens may arise and cause serious clinical conditions, or aggravation of symptoms. Typically, such reactions have been observed within the first weeks or months of initiation of CART. Relevant examples are cytomegalovirus retinitis, generalised and/or focal mycobacterial infections and pneumonia caused by Pneumocystis jiroveci (formerly known as Pneumocystis carinii). Any inflammatory symptoms should be evaluated and treatment instituted when necessary.
Autoimmune disorders (such as Graves' disease and autoimmune hepatitis) have also been reported to occur in the setting of immune reactivation: however, the reported time to onset is more variable and these events can occur many months after initiation of treatment.
Antacids
Co-administration of raltegravir with aluminium and magnesium antacids resulted in reduced raltegravir plasma levels. Co-administration of raltegravir with aluminium and/or magnesium antacids is not recommended (see section 4.5).
Rifampicin
Caution should be used when co-administering raltegravir with strong inducers of uridine diphosphate glucuronosyltransferase (UGT) 1A1 (e.g., rifampicin). Rifampicin reduces plasma levels of raltegravir; the impact on the efficacy of raltegravir is unknown. However, if co-administration with rifampicin is unavoidable, a doubling of the dose of raltegravir can be considered in adults. There are no data to guide co-administration of raltegravir with rifampicin in patients below 18 years of age (see section 4.5).
Myopathy and rhabdomyolysis
Myopathy and rhabdomyolysis have been reported. Use with caution in patients who have had myopathy or rhabdomyolysis in the past or have any predisposing issues including other medicinal products associated with these conditions (see section 4.8).
Severe skin and hypersensitivity reaction Severe, potentially life-threatening, and fatal skin reactions have been reported in patients taking raltegravir, in most cases concomitantly with other medicinal products associated with these reactions. These include cases of Stevens-Johnson syndrome and toxic epidermal necrolysis. Hypersensitivity reactions have also been reported and were characterised by rash, constitutional findings, and sometimes, organ dysfunction, including hepatic failure. Discontinue raltegravir and other suspect agents immediately if signs or symptoms of severe skin reactions or hypersensitivity reactions develop (including, but not limited to, severe rash or rash accompanied by fever, general malaise, fatigue, muscle or joint aches, blisters, oral lesions, conjunctivitis, facial oedema, hepatitis, eosinophilia, angioedema). Clinical status including liver aminotransferases should be monitored and appropriate therapy initiated. Delay in stopping raltegravir treatment or other suspect agents after the onset of severe rash may result in a life-threatening reaction.
Rash
Rash occurred more commonly in treatment-experienced patients receiving regimens containing raltegravir and darunavir compared to patients receiving raltegravir without darunavir or darunavir without raltegravir (see section 4.8).
Fructose / Sucrose
ISENTRESS granules for oral suspension contain fructose, sorbitol and sucrose. Patients with rare hereditary problems of fructose intolerance glucosegalactose malabsorption or sucrase-isomaltase insufficiency should not take this medicine.
Interaction with other medicinal products and other forms of interaction
In vitro studies indicate that raltegravir is not a substrate of cytochrome P450 (CYP) enzymes, does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 or CYP3A, does not inhibit UDP glucuronosyltransferases (UGTs) 1A1 and 2B7, does not induce CYP3A4 and does not inhibit P-glycoprotein-mediated transport. Based on these data, raltegravir is not expected to affect the pharmacokinetics of medicinal products that are substrates of these enzymes or P-glycoprotein.
Based on in vitro and in vivo studies, raltegravir is eliminated mainly by metabolism via a UGT1A1-mediated glucuronidation pathway.
Considerable inter- and intra-individual variability was observed in the pharmacokinetics of raltegravir.
Effect of raltegravir on the pharmacokinetics of other medicinal products
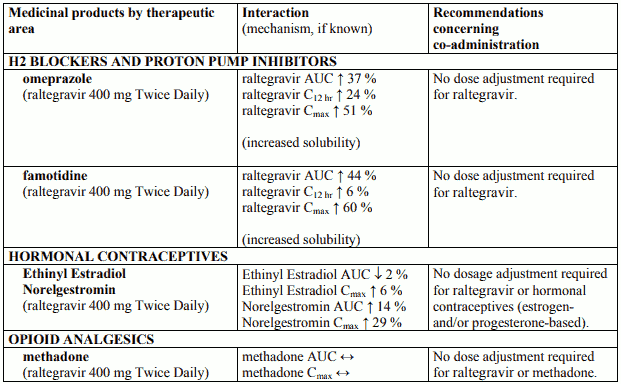
In interaction studies, raltegravir did not have a clinically meaningful effect on the pharmacokinetics of etravirine, maraviroc, tenofovir disoproxil fumarate, hormonal contraceptives, methadone, midazolam or boceprevir.
In some studies, co-administration of raltegravir with darunavir resulted in a modest decrease in darunavir plasma concentrations; the mechanism for this effect is unknown. However, the effect of raltegravir on darunavir plasma concentrations does not appear to be clinically meaningful.
Effect of other medicinal products on the pharmacokinetics of raltegravir
Given that raltegravir is metabolised primarily via UGT1A1, caution should be used when co-administering raltegravir with strong inducers of UGT1A1 (e.g., rifampicin). Rifampicin reduces plasma levels of raltegravir; the impact on the efficacy of raltegravir is unknown. However, if co-administration with rifampicin is unavoidable, a doubling of the dose of raltegravir can be considered in adults. There are no data to guide co-administration of raltegravir with rifampicin in patients below 18 years of age (see section 4.4). The impact of other strong inducers of drug metabolizing enzymes, such as phenytoin and phenobarbital, on UGT1A1 is unknown. Less potent inducers (e.g., efavirenz, nevirapine, etravirine, rifabutin, glucocorticoids, St. John's wort, pioglitazone) may be used with the recommended dose of raltegravir.
Co-administration of raltegravir with medicinal products that are known to be potent UGT1A1 inhibitors (e.g., atazanavir) may increase plasma levels of raltegravir. Less potent UGT1A1 inhibitors (e.g., indinavir, saquinavir) may also increase plasma levels of raltegravir, but to a lesser extent compared with atazanavir. In addition, tenofovir disoproxil fumarate may increase plasma levels of raltegravir, however, the mechanism for this effect is unknown (see Table 3). From the clinical trials, a large proportion of patients used atazanavir and / or tenofovir disoproxil fumarate, both agents that result in increases in raltegravir plasma levels, in the optimised background regimens. The safety profile observed in patients who used atazanavir and / or tenofovir disoproxil fumarate was generally similar to the safety profile of patients who did not use these agents. Therefore, no dose adjustment is required.
Co-administration of raltegravir with antacids containing divalent metal cations may reduce raltegravir absorption by chelation, resulting in a decrease of raltegravir plasma levels. Taking an aluminium and magnesium antacid within 6 hours of raltegravir administration significantly decreased raltegravir plasma levels. Therefore, co-administration of raltegravir with aluminium and/or magnesium containing antacids is not recommended. Co-administration of raltegravir with a calcium carbonate antacid decreased raltegravir plasma levels; however, this interaction is not considered clinically meaningful. Therefore, when raltegravir is co-administered with calcium carbonate containing antacids no dose adjustment is required. Co-administration of raltegravir with other agents that increase gastric pH (e.g., omeprazole and famotidine) may increase the rate of raltegravir absorption and result in increased plasma levels of raltegravir (see Table 3). Safety profiles in the subgroup of patients in Phase III trials taking proton pump inhibitors or H2 antagonists were comparable with those who were not taking these antacids. Therefore, no dose adjustment is required with use of proton pump inhibitors or H2 antagonists.
All interaction studies were performed in adults.
Table 3. Pharmacokinetic Interaction Data:
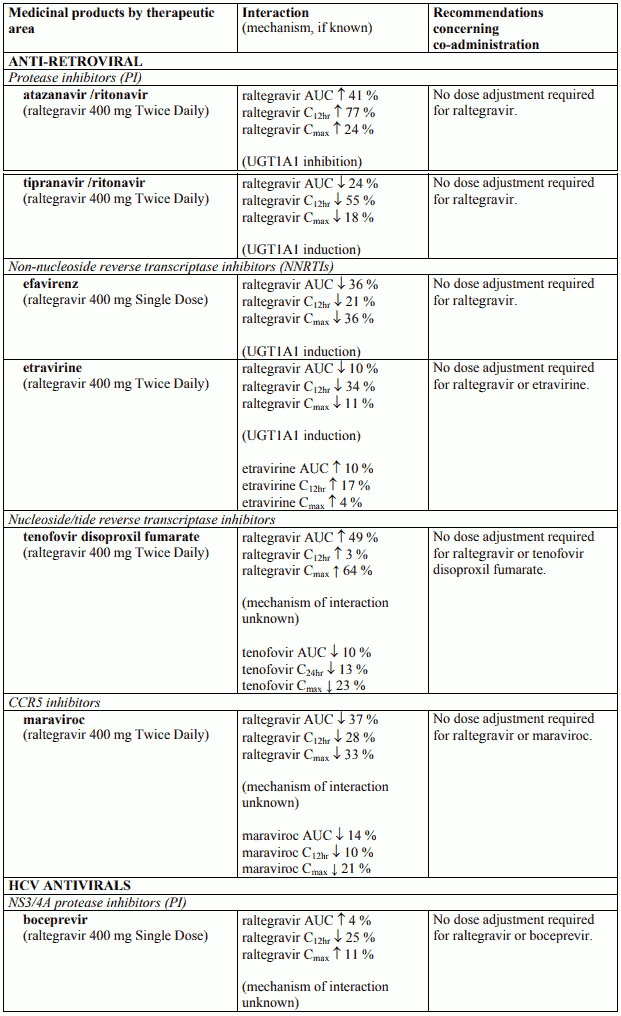

Pregnancy and lactation
Pregnancy
There are no data for the use of raltegravir granules for oral suspension in pregnant women. A moderate amount of data on pregnant women (between 300 – 1,000 pregnancy outcomes from first trimester exposure) indicate no malformative or feto/neonatal toxicity of raltegravir 400 mg film-coated tablets twice daily. Animal studies have shown reproductive toxicity (see section 5.3). Raltegravir granules for oral suspension should be used during pregnancy only if the expected benefit justifies the potential risk to the foetus. See section 4.2 for dosing recommendations.
Anti-retroviral Pregnancy Registry
To monitor maternal-foetal outcomes in patients inadvertently administered raltegravir while pregnant, an Anti-retroviral Pregnancy Registry has been established. Physicians are encouraged to register patients in this registry.
As a general rule, when deciding to use antiretroviral agents for the treatment of HIV infection in pregnant women and consequently for reducing the risk of HIV vertical transmission to the newborn, the animal data as well as the clinical experience in pregnant women should be taken into account in order to characterise the safety for the foetus.
Breast-feeding
It is unknown whether raltegravir/metabolites are excreted in human milk. Available pharmacodynamics/toxicological data in animals have shown excretion of raltegravir/metabolites in milk (for details see section 5.3).
A risk to the newborns/infants cannot be excluded.
Raltegravir should not be used during breast-feeding. As a general rule, it is recommended that mothers infected by HIV do not breast-feed their babies in order to avoid transmission of HIV.
Fertility
No effect on fertility was seen in male and female rats at doses up to 600 mg/kg/day which resulted in 3-fold exposure above the exposure at the recommended human dose.
Effects on ability to drive and use machines
Dizziness has been reported in some patients during treatment with regimens containing raltegravir. Dizziness may influence some patients' ability to drive and use machines (see section 4.8).
Undesirable effects
Summary of the safety profile
In randomised clinical trials raltegravir 400 mg twice daily was administered in combination with fixed or optimised background treatment regimens to treatment-naïve (N=547) and treatmentexperienced (N=462) adults for up to 96 weeks. A further 531 treatment-naïve adults have received raltegravir 1,200 mg once daily with emtricitabine and tenofovir disoproxil fumarate for up to 96 weeks. See section 5.1.
The most frequently reported adverse reactions during treatment were headache, nausea and abdominal pain. The most frequently reported serious adverse reactions were immune reconstitution syndrome and rash. The rates of discontinuation of raltegravir due to adverse reactions were 5% or less in clinical trials.
Rhabdomyolysis was an uncommonly reported serious adverse reaction in post-marketing use of raltegravir 400 mg twice daily.
Tabulated summary of adverse reactions
Adverse reactions considered by investigators to be causally related to raltegravir (alone or in combination with other ART), as well as adverse reactions established in post-marketing experience, are listed below by System Organ Class. Frequencies are defined as common (≥1/100 to <1/10), uncommon (≥1/1,000 to <1/100), and not known (cannot be estimated from the available data).
Adverse reactions Raltegravir (alone or in combination with other ART):
Infections and infestations
Uncommon: genital herpes, folliculitis, gastroenteritis, herpes simplex, herpes virus infection, herpes zoster, influenza, lymph node abscess, molluscum contagiosum, nasopharyngitis, upper respiratory tract infection
Neoplasms benign, malignant and unspecified (including cysts and polyps)
Uncommon: skin papilloma
Blood and lymphatic system disorders
Uncommon: anaemia, iron deficiency anaemia, lymph node pain, lymphadenopathy, neutropenia, thrombocytopenia
Immune system disorders
Uncommon: immune reconstitution syndrome, drug hypersensitivity, hypersensitivity
Metabolism and nutrition disorders
Common: decreased appetite
Uncommon: cachexia, diabetes mellitus, dyslipidaemia, hypercholesterolaemia, hyperglycaemia, hyperlipidaemia, hyperphagia, increased appetite, polydipsia, body fat disorder
Psychiatric disorders
Common: abnormal dreams, insomnia, nightmare, abnormal behaviour, depression
Uncommon: mental disorder, suicide attempt, anxiety, confusional state, depressed mood, major depression, middle insomnia, mood altered, panic attack, sleep disorder, suicidal ideation, suicidal behaviour (particularly in patients with a pre-existing history of psychiatric illness)
Nervous system disorders
Common: dizziness, headache, psychomotor hyperactivity
Uncommon: amnesia, carpal tunnel syndrome, cognitive disorder, disturbance in attention, dizziness postural, dysgeusia, hypersomnia, hypoaesthesia, lethargy, memory impairment, migraine, neuropathy peripheral, paraesthesia, somnolence, tension headache, tremor, poor quality sleep
Eye disorders
Uncommon: visual impairment
Ear and labyrinth disorders
Common: Uncommon vertigo tinnitus
Cardiac disorders
Uncommon: palpitations, sinus bradycardia, ventricular extrasystoles
Vascular disorders
Uncommon: hot flush, hypertension Respiratory, thoracic and mediastinal disorders
Uncommon: dysphonia, epistaxis, nasal congestion
Gastrointestinal disorders
Common: abdominal distention, abdominal pain, diarrhoea, flatulence, nausea, vomiting, dyspepsia
Uncommon: gastritis, abdominal discomfort, abdominal pain upper, abdominal tenderness, anorectal discomfort, constipation, dry mouth, epigastric discomfort, erosive duodenitis, eructation, gastroesophageal reflux disease, gingivitis, glossitis, odynophagia, pancreatitis acute, peptic ulcer, rectal haemorrhage
Hepato-biliary disorders
Uncommon: hepatitis, hepatic steatosis, hepatitis alcoholic, hepatic failure
Skin and subcutaneous tissue disorders
Common: rash
Uncommon: acne, alopecia, dermatitis acneiforme, dry skin, erythema, facial wasting, hyperhidrosis, lipoatrophy, lipodystrophy acquired, lipohypertrophy, night sweats, prurigo, pruritus, pruritus generalised, rash macular, rash maculopapular, rash pruritic, skin lesion, urticaria, xeroderma, Stevens Johnson syndrome, drug rash with eosinophilia and systemic symptoms (DRESS)
Musculoskeletal and connective tissue disorders
Uncommon: arthralgia, arthritis, back pain, flank pain, musculoskeletal pain, myalgia, neck pain, osteopenia, pain in extremity, tendonitis, rhabdomyolysis
Renal and urinary disorders
__Uncommon :__renal failure, nephritis, nephrolithiasis, nocturia, renal cyst, renal impairment, tubulointerstitial nephritis
Reproductive system and breast disorders
Uncommon: erectile dysfunction, gynaecomastia, menopausal symptoms
General disorders and administration site conditions
Common: asthenia, fatigue, pyrexia Uncommon chest discomfort, chills, face oedema, fat tissue increased, feeling jittery, malaise, submandibular mass, oedema peripheral, pain
Investigations
Common: alanine aminotransferase increased, atypical lymphocytes, aspartate aminotransferase increased, blood triglycerides increased, lipase increased, blood pancreatic amylase increased
Uncommon: absolute neutrophil count decreased, alkaline phosphatase increased, blood albumin decreased, blood amylase increased, blood bilirubin increased, blood cholesterol increased, blood creatinine increased, blood glucose increased, blood urea nitrogen increased, creatine phosphokinase increased, fasting blood glucose increased, glucose urine present, high density lipoprotein increased, international normalised ratio increased, low density lipoprotein increased, platelet count decreased, red blood cells urine positive, waist circumference increased, weight increased, white blood cell count decreased
Injury, poisoning and procedural complications
Uncommon: accidental overdose
Description of selected adverse reactions
In studies of raltegravir 400 mg twice daily, cancers were reported in treatment-experienced and treatment-naïve patients who initiated raltegravir in conjunction with other antiretroviral agents. The types and rates of specific cancers were those expected in a highly immunodeficient population. The risk of developing cancer in these studies was similar in the groups receiving raltegravir and in the groups receiving comparators.
Grade 2-4 creatine kinase laboratory abnormalities were observed in patients treated with raltegravir. Myopathy and rhabdomyolysis have been reported. Use with caution in patients who have had myopathy or rhabdomyolysis in the past or have any predisposing issues including other medicinal products associated with these conditions (see section 4.4).
Cases of osteonecrosis have been reported, particularly in patients with generally acknowledged risk factors, advanced HIV disease or long-term exposure to combination antiretroviral therapy (CART). The frequency of this is unknown (see section 4.4).
In HIV-infected patients with severe immune deficiency at the time of initiation of combination antiretroviral therapy (CART), an inflammatory reaction to asymptomatic or residual opportunistic infections may arise. Autoimmune disorders (such as Graves' disease and autoimmune hepatitis) have also been reported; however, the reported time to onset is more variable and these events can occur many months after initiation of treatment (see section 4.4).
For each of the following clinical adverse reactions there was at least one serious occurrence: genital herpes, anaemia, immune reconstitution syndrome, depression,mental disorder, suicide attempt, gastritis, hepatitis, renal failure, accidental overdose.
In clinical studies of treatment-experienced patients, rash, irrespective of causality, was more commonly observed with regimens containing raltegravir and darunavir compared to those containing raltegravir without darunavir or darunavir without raltegravir. Rash considered by the investigator to be drug-related occurred at similar rates. The exposure-adjusted rates of rash (all causality) were 10.9, 4.2, and 3.8 per 100 patient-years (PYR), respectively; and for drug-related rash were 2.4, 1.1, and 2.3 per 100 PYR, respectively. The rashes observed in clinical studies were mild to moderate in severity and did not result in discontinuation of therapy (see section 4.4).
Patients co-infected with hepatitis B and/or hepatitis C virus
In clinical trials, there were 79 patients co-infected with hepatitis B, 84 co-infected with hepatitis C, and 8 patients co-infected with hepatitis B and C who were treated with raltegravir in combination with other agents for HIV-1. In general, the safety profile of raltegravir in patients with hepatitis B and/or hepatitis C virus co-infection was similar to that in patients without hepatitis B and/or hepatitis C virus co-infection, although the rates of AST and ALT abnormalities were somewhat higher in the subgroup co-infected with hepatitis B and/or hepatitis C virus.
At 96-weeks, in treatment-experienced patients, Grade 2 or higher laboratory abnormalities that represent a worsening Grade from baseline of AST, ALT or total bilirubin occurred in 29 %, 34 % and 13 %, respectively, of co-infected patients treated with raltegravir as compared to 11 %, 10 % and 9 % of all other patients treated with raltegravir. At 240-weeks, in treatment-naïve patients, Grade 2 or higher laboratory abnormalities that represent a worsening Grade from baseline of AST, ALT or total bilirubin occurred in 22 %, 44 % and 17 %, respectively, of co-infected patients treated with raltegravir as compared to 13 %, 13 % and 5 % of all other patients treated with raltegravir.
Paediatric population
ISENTRESS 600 mg tablet formulation has not been studied in paediatric patients (see section 4.2).
Children and adolescents 2 to 18 years of age
Raltegravir twice daily has been studied in 126 antiretroviral treatment-experienced HIV-1 infected children and adolescents 2 to 18 years of age, in combination with other antiretroviral agents in IMPAACT P1066 (see sections 5.1 and 5.2). Of the 126 patients, 96 received the recommended dose of raltegravir twice daily.
In these 96 children and adolescents, frequency, type and severity of drug related adverse reactions through Week 48 were comparable to those observed in adults.
One patient experienced drug related clinical adverse reactions of Grade 3 psychomotor hyperactivity, abnormal behaviour and insomnia; one patient experienced a Grade 2 serious drug related allergic rash.
One patient experienced drug related laboratory abnormalities, Grade 4 AST and Grade 3 ALT, which were considered serious.
Infants and toddlers 4 weeks to less than 2 years of age
Raltegravir twice daily has also been studied in 26 HIV-1 infected infants and toddlers 4 weeks to less than 2 years of age, in combination with other antiretroviral agents in IMPAACT P1066 (see sections 5.1 and 5.2).
In these 26 infants and toddlers, the frequency, type and severity of drug related adverse reactions through Week 48 were comparable to those observed in adults.
One patient experienced a Grade 3 serious drug related allergic rash that resulted in treatment discontinuation.
HIV-1 Exposed Neonates
In IMPAACT P1110 (see section 5.2) eligible infants were at least 37 weeks gestation and at least 2 kg in weight. Sixteen (16) neonates received 2 doses of ISENTRESS in first 2 weeks of life, and 26 neonates received 6 weeks of daily dosing; all were followed for 24 weeks. There were no drug related clinical adverse experiences and three drug-related laboratory adverse experiences (one a transient Grade 4 neutropenia in a subject receiving zidovudine containing prevention of mother to child transmission (PMTCT), and two bilirubin elevations (one each, Grade 1 and Grade 2) considered non-serious and not requiring specific therapy).
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed in Appendix V.
Incompatibilities
Not applicable.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.