ITOVEBI Film-coated tablet Ref.[115587] Active ingredients: Inavolisib
Source: European Medicines Agency (EU) Revision Year: 2025 Publisher: Roche Registration GmbH, Emil-Barell-Strasse 1, 79639 Grenzach-Wyhlen, Germany
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic agents, PI3K inhibitors
ATC Code: not yet assigned
Mechanism of action
Inavolisib is an inhibitor of the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) catalytic subunit alpha isoform protein (p110α; encoded by the PIK3CA gene). In addition, inavolisib promotes the degradation of mutated p110α (mutant degrader). The PI3K signalling pathway is commonly dysregulated in HR-positive breast cancer, often due to activating PIK3CA mutations. With its dual mechanism of action, inavolisib inhibits the activity of downstream PI3K pathway targets, including AKT, resulting in reduced cellular proliferation and induction of apoptosis in PIK3CA-mutated breast cancer cell lines.
Clinical efficacy and safety
Locally advanced or metastatic breast cancer
The patients in this setting, based on data from the INAVO120 study, are defined as endocrine-resistant patients (disease recurrence on or within 12 months of adjuvant endocrine treatment completion) who have not received prior treatment for their locally advanced or metastatic disease.
INAVO120
The efficacy of Itovebi in combination with palbociclib and fulvestrant was evaluated in a Phase 3, randomised, double-blind, placebo-controlled study in adult patients with PIK3CA-mutated, HR-positive, HER2-negative, locally advanced or metastatic breast cancer whose disease progressed during or within 12 months of completing adjuvant endocrine therapy (endocrine-resistant) and who have not received prior systemic therapy for locally advanced or metastatic disease. The study included patients who received prior (neo)adjuvant endocrine therapy including a CDK4/6 inhibitor if the progression event was >12 months since completion of the CDK4/6 inhibitor portion of (neo)adjuvant therapy, and who had HbA1C <6% and fasting blood glucose <126 mg/dL. The study excluded patients with Type 1 diabetes mellitus or Type 2 diabetes mellitus requiring ongoing anti-hyperglycaemic therapy at the start of study treatment, patients who received prior treatment with fulvestrant (except as part of neoadjuvant therapy with treatment duration ≤6 months), and patients with known and untreated, or active CNS metastases (progressing or requiring anticonvulsants or corticosteroids for symptomatic control).
PIK3CA mutation status was prospectively determined through testing of plasma-derived circulating tumour DNA (ctDNA) using a next-generation sequencing (NGS) assay (FoundationOne Liquid CDx assay or PredicineCARETM) performed at a central laboratory (87.4%), or in local laboratories (12.6%) using various validated polymerase chain reaction (PCR) or NGS assays on tumour tissue or plasma. The following PIK3CA mutations at the indicated amino acid positions were eligible for inclusion: H1047D/I/L/N/P/Q/R/T/Y, G1049A/C/D/R/S, E545A/D/G/K/L/Q/R/V, E453A/D/G/K/Q/V, E542A/D/G/K/Q/R/V, K111N/R/E, Q546E/H/K/L/P/R, G106A/D/R/S/V, N345D/H/I/K/S/T/Y, G118D, C420R, R88Q, and M1043I/T/V. At least one eligible PIK3CA mutation was identified in at least one of these amino acid positions in each of the enrolled patient specimens. Based on results from the central FoundationOne Liquid CDx assay, the most common PIK3CA alterations were short variants at amino acids H1047 (n=115, 42.6%), E545 (n=58, 21.5%), and E542 (n=39, 14.4%). There were 25 patients whose specimens harboured more than one PIK3CA alterations (i.e., multiple PIK3CA mutations), and 33 with less common PIK3CA alterations.
A total of 325 patients were randomised 1:1 to receive either Itovebi 9 mg (n=161) or placebo (n=164) orally once daily, in combination with palbociclib and fulvestrant, until disease progression or unacceptable toxicity. In addition, pre/perimenopausal women and men received an LHRH agonist throughout therapy. Randomisation was stratified by presence of visceral disease (yes or no), endocrine resistance (primary or secondary), and geographic region (North America/Western Europe, Asia, other).
The baseline demographic and disease characteristics were: median age 54 years (range: 27 to 79 years, 18.2% were ≥65 years of age); 98.2% female; 38.2% pre/perimenopausal; 58.8% White, 38.2% Asian, 2.5% unknown, 0.6% Black or African American; 6.2% Hispanic or Latino; and Eastern Cooperative Oncology Group (ECOG) performance status of 0 (63.4%) or 1 (36.3%). Tamoxifen (56.9%) and aromatase inhibitors (50.2%) were the most commonly used adjuvant endocrine therapies. Three (0.9%) patients received prior CDK4/6 inhibitor therapy. The demographics and baseline disease characteristics were balanced and comparable between study arms.
The primary efficacy outcome measure was investigator (INV)-assessed progression-free survival (PFS) per Response Evaluation Criteria in Solid Tumours (RECIST) version 1.1. The secondary efficacy outcome measures included overall survival (OS), objective response rate (ORR), best overall response (BOR), clinical benefit rate (CBR), duration of response (DOR), and time to confirmed deterioration (TTCD) in pain, physical function, role function, and global health status/health-related quality of life (HRQoL).
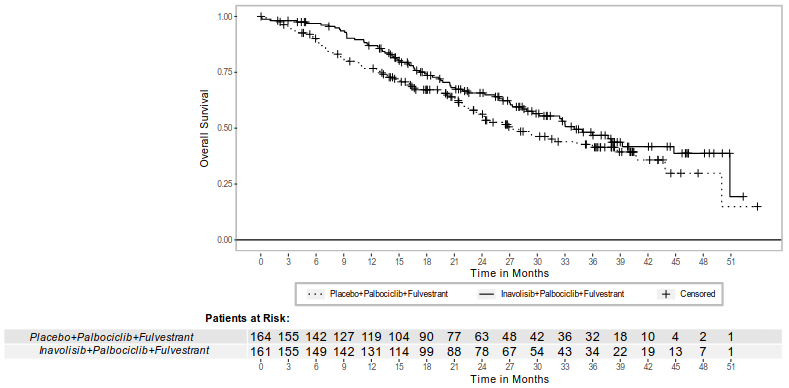
Efficacy results are summarised in Table 7, Figure 1, and Figure 2. INV-assessed PFS results were supported by consistent results from blinded independent central review (BICR) assessment.
Table 7. Efficacy results in patients with locally advanced or metastatic breast cancer in INAVO120:
| Efficacy endpoint | Itovebi + palbociclib + fulvestrant N=161 | Placebo + palbociclib + fulvestrant N=164 |
|---|---|---|
| INV-assessed progression-free survivala | ||
| Patients with event, n (%) | 82 (50.9) | 113 (68.9) |
| Median, months (95% CI) | 15 (11.3, 20.5) | 7.3 (5.6, 9.3) |
| Hazard ratio (95% CI) | 0.43 (0.32, 0.59) | |
| p-value | <0.0001 | |
| Overall survivalb,c | ||
| Patients with event, n (%) | 72 (44.7) | 82 (50) |
| Median, months (95% CI) | 34 (28.4, 44.8) | 27 (22.8, 38.7) |
| Hazard ratio (95% CI) | 0.67 (0.48, 0.94) | |
| p-value | 0.0190 | |
| Objective response rateb,d | ||
| Patients with CR or PR, n (%) | 101 (62.7) | 46 (28) |
| 95% CI | (54.8, 70.2) | (21.3, 35.6) |
| p-value | <0.0001 | |
| Duration of responseb | ||
| Median DOR, months (95% CI) | 19.2 (14.7, 28.3) | 11.1 (8.5, 20.2) |
CI = confidence interval; CR = complete response; PR = partial response
a Per RECIST version 1.1. Based on primary analysis (clinical cutoff date: 29 September 2023).
b Based on final overall survival analysis (clinical cutoff date: 15 November 2024).
c The prespecified boundary for statistical significance was p<0.0469.
d Per RECIST version 1.1. ORR is defined as the proportion of patients with a CR or PR on two consecutive occasions ≥4 weeks apart, as determined by the investigator.
Figure 1. INV-assessed progression-free survival in patients with locally advanced or metastatic breast cancer in INAVO120:
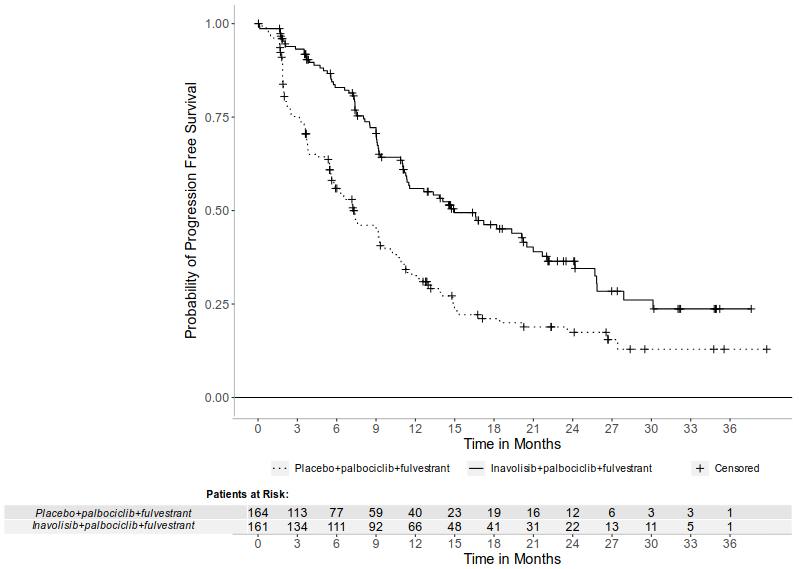
Figure 2. Overall survival in patients with locally advanced or metastatic breast cancer in INAVO120:
Paediatric population
The European Medical Agency has waived the obligation to submit the results of studies with Itovebi in all subsets of the paediatric population in breast cancer (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
The pharmacokinetics of inavolisib were characterised in healthy subjects and in patients with locally advanced or metastatic PIK3CA-mutated solid tumours, including breast cancer, under an oral dosing regimen ranging from 6 mg to 12 mg daily and in healthy subjects at 9 mg single dose.
Inavolisib pharmacokinetics are presented as geometric mean (geometric coefficient of variation [geo CV]%) following administration of the approved recommended dosage unless otherwise specified. Based on population pharmacokinetics analysis, the inavolisib steady-state AUC was 1 019 h*ng/mL (29%) and Cmax was 67 ng/mL (28%). Steady-state concentrations were predicted to be attained by day 5.
With 9 mg once daily dosing, the geometric mean accumulation ratio was about 2-fold.
Absorption
The time to maximum plasma concentration (Tmax) was reached after a median of 3 hours (range: 0.5 to 4 hours) at steady state following 9 mg daily dosing of inavolisib, under fasted conditions.
The absolute oral bioavailability of inavolisib was 76%.
Food effect
No clinically significant effect of food on inavolisib exposure was observed. The geometric mean ratio (GMR) (90% CI) for AUC0-24 comparing the fed to the fasted state was 0.895 (0.737–1.09) after a single dose and 0.876 (0.701–1.09) at steady state. The GMR (90% CI) for Cmax comparing the fed to the fasted state was 0.925 (0.748–1.14) after a single dose and 0.910 (0.712–1.16) at steady state.
Distribution
Plasma protein binding of inavolisib in humans is 37% and did not appear to be concentration-dependent over the concentration range tested (0.1–10 μM). In humans, the estimated steady state oral volume of distribution is 155 L (26%) based on population pharmacokinetics analysis.
Biotransformation
Following oral administration of a single radio-labeled 9 mg dose of inavolisib to healthy subjects, parent drug was the most prominent drug-related compound in plasma and urine. Hydrolysis was the major metabolic pathway. No specific hydrolysis enzymes involved in the metabolism of inavolisib were identified.
Elimination
Following oral administration of a single radio-labeled 9 mg dose of inavolisib to healthy subjects, 48.5% of the administered dose was recovered in urine (40.4% unchanged) and 48% in faeces (10.8% unchanged).
In clinical studies, based on population pharmacokinetics analysis, the geometric mean of the individual elimination half-life estimates for inavolisib was 15 hours (24%) following a single 9 mg dose. The estimated total clearance of inavolisib is 8.8 L/hr (29%).
Linearity/non-linearity
Limited data suggest dose proportionality within the dose range tested (6 to 12 mg) for single-dose Cmax and AUC0-24 and steady-state AUC0-24; however, for steady-state Cmax, the data suggest non- proportionality.
Drug-drug interactions
Clinical study results indicated that the predominant metabolites of inavolisib are not mediated by CYP enzymes, suggesting a low likelihood of clinically relevant interactions between inavolisib and CYP inhibitors or inducers. Moreover, in vitro results indicated that inavolisib does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP2D6 enzymes.
In vitro studies have shown that inavolisib does not appear to have the potential to inhibit any of the relevant drug transporters tested. Furthermore, inavolisib is a substrate of P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP) in vitro. However, based on the overall pharmacokinetic characteristics of inavolisib, inhibitors or inducers of P-gp and/or BCRP are not expected to cause a clinically relevant drug-drug interaction with inavolisib.
Special populations
Elderly
No clinically relevant differences in inavolisib pharmacokinetics were noted between patients 65 years of age and older and those under 65 years based on population pharmacokinetic analysis. Of the 162 patients who received Itovebi in the INAVO120 study, 24 patients were ≥65 years of age.
Renal impairment
Population pharmacokinetic analyses indicated that mild renal impairment is not a clinically relevant covariate on inavolisib exposure. The pharmacokinetics of inavolisib in patients with mild renal impairment (eGFR 60 to <90 mL/min) were similar to those in patients with normal renal function. Inavolisib AUC and Cmax were 73% and 11% higher in patients with moderate renal impairment compared to patients with normal renal function (eGFR ≥90 mL/min), respectively. The effect of severe renal impairment on inavolisib pharmacokinetics has not been established.
Hepatic impairment
Population pharmacokinetic analyses indicated that mild hepatic impairment is not a clinically relevant covariate on inavolisib exposure. The pharmacokinetics of inavolisib in patients with mild hepatic impairment (total bilirubin > ULN to ≤ 1.5 × ULN or AST > ULN and total bilirubin ≤ ULN) were similar to those in patients with normal hepatic function. The effect of moderate to severe hepatic impairment on inavolisib pharmacokinetics has not been studied.
5.3. Preclinical safety data
Genotoxicity
Inavolisib was not mutagenic in the bacterial mutagenesis assay.
Inavolisib showed clastogenicity in vitro; however, there was no evidence of inavolisib-induced in vivo genotoxicity (clastogenicity, aneugenicity, or DNA damage) in the micronucleus and comet study in rats at doses up to a maximum tolerated dose (MTD) of 16 times the exposure at a clinical dose of 9 mg.
Carcinogenicity
No carcinogenicity studies with inavolisib have been conducted.
Developmental toxicity
An embryo-foetal development study in Sprague-Dawley rats identified inavolisib-related dose-dependent effects on embryo-foetal development that included decreases in foetal body weight and placental weight, post-implantation loss, lower foetal viability, and teratogenicity (foetal external, visceral, and skeletal malformations), with the maternal exposure at NOAEL being 0.2 times the exposure at a clinical dose of 9 mg.
Fertility
No dedicated fertility studies with inavolisib have been conducted.
In male rats, dose-dependent atrophy of the prostate and seminal vesicle and decreased organ weights without microscopic correlate in the epididymis and testis were observed (at ≥ NOAEL of 0.4 times the exposure at a clinical dose of 9 mg). These findings were reversible. In male dogs, focal inspissation of seminiferous tubule contents and multinucleated spermatids in the testis and epithelial degeneration/necrosis in the epididymis were observed following 4 weeks of dosing (at ≥2 times the exposure at a clinical dose of 9 mg). Following 3 months of dosing at up to 1.2 times the exposure at a clinical dose of 9 mg, a reversible decrease in total sperm count was observed with a no observed adverse effect level (NOAEL) of 0.4 times the exposure at a clinical dose of 9 mg but there were no inavolisib-related microscopic findings in the testes or epididymides or effects on sperm concentration, motility, or morphology.
In female rats, minimal to mild atrophy in the uterus and vagina, decreased ovarian follicles, and findings suggestive of an interruption/alteration of the oestrus cycle were observed (at ≥1.2 times the exposure at a clinical dose of 9 mg), with a NOAEL of 0.5 times the exposure at a clinical dose of 9 mg. These findings were not observed following the recovery period in the 4-week toxicity study. Recovery was not assessed in the 3-month study in rats.
Other
Adverse reactions not observed in clinical studies, but seen in animals at exposure levels similar to clinical exposure levels and with possible relevance to clinical use, included inflammation in dogs and eye lens degeneration in rats. The inflammation is consistent with the anticipated pharmacologic effects of PI3K inhibition, was generally dose-dependent and reversible. Minimal lens fiber degeneration observed in some rats (at ≥3.6 times the exposure at a clinical dose of 9 mg) was considered irreversible.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.