JADENU Film-coated tablet / Granules Ref.[10853] Active ingredients: Deferasirox
Source: FDA, National Drug Code (US) Revision Year: 2020
12.1. Mechanism of Action
JADENU (deferasirox) is an orally active chelator that is selective for iron (as Fe3+). It is a tridentate ligand that binds iron with high affinity in a 2:1 ratio. Although deferasirox has very low affinity for zinc and copper, there are variable decreases in the serum concentration of these trace metals after the administration of deferasirox. The clinical significance of these decreases is uncertain.
12.2. Pharmacodynamics
Pharmacodynamic effects tested in an iron balance metabolic study with the tablet for oral suspension formulation showed that deferasirox (10, 20, and 40 mg per kg per day) was able to induce a mean net iron excretion (0.119, 0.329, and 0.445 mg Fe/kg body weight per day, respectively) within the clinically relevant range (0.1 to 0.5 mg per kg per day). Iron excretion was predominantly fecal.
An analysis of pooled pediatric clinical trial data found a statistically significant relationship between exposure and the probability of renal toxicity (increase in serum creatinine and urinary protein), resulting in a decrease in renal function. Decreases in renal function resulted in an increase in deferasirox exposure which may increase the probability of renal toxicity.
Cardiac Electrophysiology
At the maximum approved recommended dose, deferasirox does not prolong the QT interval to any clinically relevant extent.
12.3. Pharmacokinetics
Absorption
Based on studies in patients with the tablet for oral suspension, deferasirox is absorbed following oral administration with median times to maximum plasma concentration (Tmax) of about 1.5 to 4 hours. In healthy subjects, JADENU showed comparable Tmax. The maximal concentrations (Cmax) and area under the curve (AUC0-24h, AUCτ) of deferasirox increase approximately linearly with dose after both single administration and under steady-state conditions. Exposure to deferasirox increased by an accumulation factor of 1.3 to 2.3 after multiple doses with the tablet for oral suspension formulation.
Tablets
The absolute bioavailability [as measured by area under the curve over time to infinity (AUCinf)] of deferasirox tablets for oral suspension is 70% compared to an intravenous dose. The bioavailability (as measured by AUCinf) of JADENU tablets was 36% greater than with deferasirox tablets for oral suspension. After strength-adjustment, the mean AUCinf of JADENU tablets (i.e., 360 mg strength) was similar to that of deferasirox tablets for oral suspension (i.e., 500 mg strength) under fasting conditions; however the mean Cmax was increased by 30%. The 30% increase in Cmax observed with JADENU tablets is not clinically meaningful.
The administration of JADENU tablets with a light meal (approximately 250 calories with fat content less than 7% of total calories) indicated that the AUCinf and Cmax were similar to that under fasting conditions. The administration of JADENU tablets with a high-fat meal (approximately 1,000 calories with fat content greater than 50% of total calories), increased AUCinf by 18% and Cmax by 29% compared to that under fasting conditions [see Dosage and Administration (2.3)].
Granules
The bioavailability (as measured by AUCinf) of JADENU Sprinkle granules was 52% greater than with deferasirox tablets for oral suspension. After strength-adjustment, the mean AUCinf of the JADENU Sprinkle granules (i.e., 4 x 90 mg strength) was similar to that of deferasirox tablets for oral suspension (i.e., 500 mg strength) under fasting conditions; however, the mean Cmax was increased by 34%. The 34% increase in Cmax observed with JADENU Sprinkle granules is not clinically meaningful.
The administration of JADENU Sprinkle granules with a soft meal (e.g., yogurt and applesauce) or with a low-fat (approximately 450 calories with fat content approximately 30% of total calories) indicated that the AUCinf and Cmax after a low-fat meal or soft foods were similar to that under fasting conditions. The administration of JADENU Sprinkle granules with a high-fat meal (approximately 1,000 calories with fat content greater than 50% of total calories) increased AUCinf by 18% with no changes in Cmax compared to that under fasting conditions [see Dosage and Administration (2.3)].
Distribution
Deferasirox is highly (~99%) protein bound almost exclusively to serum albumin. The percentage of deferasirox confined to the blood cells was 5% in humans. The volume of distribution at steady state (Vss) of deferasirox is 14.37 ± 2.69 L in adults.
Metabolism
Glucuronidation is the main metabolic pathway for deferasirox, with subsequent biliary excretion. Deconjugation of glucuronidates in the intestine and subsequent reabsorption (enterohepatic recycling) is likely to occur. Deferasirox is mainly glucuronidated by UGT1A1 and to a lesser extent UGT1A3. CYP450-catalyzed (oxidative) metabolism of deferasirox appears to be minor in humans (about 8%). Deconjugation of glucuronide metabolites in the intestine and subsequent reabsorption (enterohepatic recycling) was confirmed in a healthy subjects study in which the administration of cholestyramine 12 g twice daily (strongly binds to deferasirox and its conjugates) 4 and 10 hours after a single dose of deferasirox resulted in a 45% decrease in deferasirox exposure (AUCinf) by interfering with the enterohepatic recycling of deferasirox.
Excretion
Deferasirox and metabolites are primarily (84% of the dose) excreted in the feces. Renal excretion of deferasirox and metabolites is minimal (8% of the dose). The mean elimination half-life (t1/2) ranged from 8 to 16 hours following oral administration.
Drug Interactions
Midazolam: The concomitant administration of deferasirox tablets for oral suspension and CYP3A4 probe substrate midazolam resulted in a decrease of midazolam Cmax by 23% and AUCinf by 17%. In the clinical setting, this effect may be more pronounced, as the study was not adequately designed to conclusively assess the potential induction of CYP3A4 by deferasirox [see Drug Interactions (7.2)].
Repaglinide: The concomitant administration of deferasirox tablets for oral suspension (30 mg per kg/day for 4 days) and the CYP2C8 probe substrate repaglinide (single dose of 0.5 mg) increased repaglinide AUCinf to 2.3-fold and Cmax of 1.6-fold [see Drug Interactions (7.3)].
Theophylline: The concomitant administration of deferasirox tablets for oral suspension (repeated dose of 30 mg per kg/day) and the CYP1A2 substrate theophylline (single dose of 120 mg) resulted in an approximate doubling of the theophylline AUCinf and elimination half-life. The single dose Cmax was not affected, but an increase in theophylline Cmax is expected to occur with chronic dosing [see Drug Interactions (7.4)].
Rifampicin: The concomitant administration of deferasirox tablets for oral suspension (single dose of 30 mg per kg) and the strong uridine diphosphate glucuronosyltransferase (UGT) inducer rifampicin (600 mg per day for 9 days) decreased deferasirox AUCinf by 44% [see Drug Interactions (7.5)].
Cholestyramine: The concomitant administration of cholestyramine after a single dose of deferasirox tablets for oral suspension decreased deferasirox AUCinf by 45% [see Drug Interactions (7.6)].
Busulfan: Concomitant administration of deferasirox and busulfan resulted in an increase of busulfan exposure (AUC).
In vitro Studies
Deferasirox inhibited human CYP2A6, CYP2D6, and CYP2C19 in vitro.
Deferasirox is not a substrate of P-glycoprotein, MRP1 or MRP2.
Pharmacokinetics in Specific Populations
Pediatric
Following oral administration of single or multiple doses, systemic exposure of adolescents and children to deferasirox was less than in adult patients. In children less than 6 years of age, systemic exposure was about 50% lower than in adults.
Sex
The apparent clearance is 17.5% lower in females compared to males.
Renal Impairment
Compared to patients with MDS and eGFR greater than 60 mL/min/1.73 m², patients with MDS and eGFR 40 to 60 mL/min/1.73 m² (n=34) had approximately 50% higher mean deferasirox trough plasma concentrations.
Hepatic Impairment
In a single dose (20 mg/kg) study in patients with varying degrees of hepatic impairment, deferasirox exposure was increased compared to patients with normal hepatic function. The average total (free and bound) AUCinf of deferasirox increased 16% in 6 patients with mild (Child-Pugh A) hepatic impairment, and 76% in 6 patients with moderate (Child-Pugh B) hepatic impairment compared to 6 patients with normal hepatic function. The impact of severe (Child-Pugh C) hepatic impairment was assessed in only 1 patient.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
A 104-week oral carcinogenicity study in Wistar rats showed no evidence of carcinogenicity from deferasirox at doses up to 60 mg/kg/day (0.7 times the MRHD on an mg/m² basis). A 26-week oral carcinogenicity study in p53 (+/-) transgenic mice has shown no evidence of carcinogenicity from deferasirox at doses up to 200 mg/kg/day (1.2 times the MRHD on a mg/m² basis) in males and 300 mg/kg/day (1.7 times the MRHD on a mg/m² basis) in females.
Deferasirox was negative in the Ames test and chromosome aberration test with human peripheral blood lymphocytes. It was positive in 1 of 3 in vivo oral rat micronucleus tests.
Deferasirox at oral doses up to 75 mg/kg/day (0.9 times the MRHD on a mg/m² basis) was found to have no adverse effect on fertility and reproductive performance of male and female rats.
14. Clinical Studies
JADENU was evaluated in healthy subjects. There are no clinical data in patients with JADENU. JADENU contains the same active ingredient as Exjade (deferasirox) tablets for oral suspension. The following information is based on clinical trials conducted with Exjade tablets for oral suspension.
Transfusional Iron Overload
The primary efficacy study, Study 1 (NCT00061750), was a multicenter, open-label, randomized, active-comparator control study to compare deferasirox tablets for oral suspension and deferoxamine in patients with beta-thalassemia and transfusional hemosiderosis. Patients greater than or equal to 2 years of age were randomized in a 1:1 ratio to receive either oral deferasirox tablets for oral suspension at starting doses of 5, 10, 20, or 30 mg per kg once daily or subcutaneous deferoxamine at starting doses of 20 to 60 mg per kg for at least 5 days per week based on LIC at baseline (2 to 3, greater than 3 to 7, greater than 7 to 14, and greater than 14 mg Fe/g dry weight). Patients randomized to deferoxamine who had LIC values less than 7 mg Fe/g dry weight were permitted to continue on their prior deferoxamine dose, even though the dose may have been higher than specified in the protocol.
Patients were to have a liver biopsy at baseline and end of study (after 12 months) for LIC. The primary efficacy endpoint was defined as a reduction in LIC of greater than or equal to 3 mg Fe/g dry weight for baseline values greater than or equal to 10 mg Fe/g dry weight, reduction of baseline values between 7 and less than 10 to less than 7 mg Fe/g dry weight, or maintenance or reduction for baseline values less than 7 mg Fe/g dry weight.
A total of 586 patients were randomized and treated, 296 with deferasirox tablets for oral suspension and 290 with deferoxamine. The mean age was 17.1 years (range, 2 to 53 years); 52% were females and 88% were Caucasian. The primary efficacy population consisted of 553 patients (deferasirox tablets for oral suspension n=276; deferoxamine n=277) who had LIC evaluated at baseline and 12 months or discontinued due to an adverse reaction. The percentage of patients achieving the primary endpoint was 52.9% for deferasirox tablets for oral suspension and 66.4% for deferoxamine. The relative efficacy of deferasirox to deferoxamine cannot be determined from this study.
In patients who had an LIC at baseline and at end of study, the mean change in LIC was -2.4 mg Fe/g dry weight in patients treated with deferasirox tablets for oral suspension and -2.9 mg Fe/g dry weight in patients treated with deferoxamine.
Reduction of LIC and serum ferritin was observed with deferasirox tablet for oral suspension doses of 20 to 30 mg per kg per day. Deferasirox tablets for oral suspension doses below 20 mg per kg per day failed to provide consistent lowering of LIC and serum ferritin levels (Figure 1). Therefore, a starting dose of 20 mg per kg per day is recommended [see Dosage and Administration (2.1)].
Figure 1. Chang es in Liver Iron Concentration and Serum Ferritin Following Deferasirox Tablets for Oral Suspension (5 to 30 mg per kg per day) in Study 1:
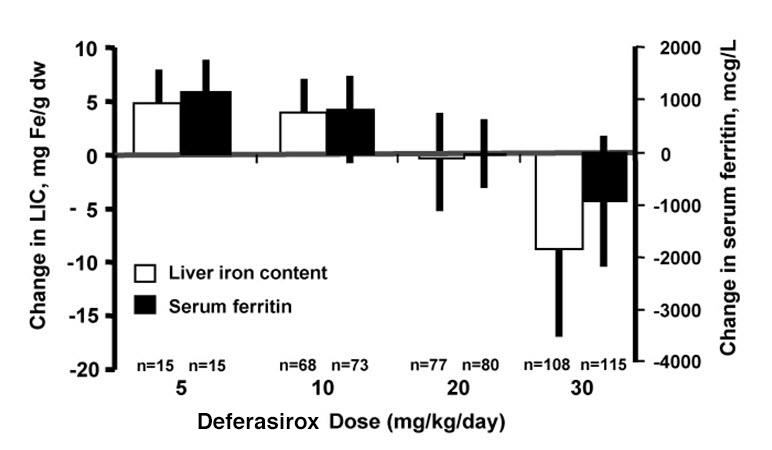
Study 2 (NCT00061763) was an open-label, noncomparative trial of efficacy and safety of deferasirox tablets for oral suspension given for 1 year to patients with chronic anemias and transfusional hemosiderosis. Similar to Study 1, patients received 5, 10, 20, or 30 mg per kg per day of deferasirox tablets for oral suspension based on baseline LIC.
A total of 184 patients were treated in this study: 85 patients with beta-thalassemia and 99 patients with other congenital or acquired anemias (myelodysplastic syndromes, n=47; Diamond-Blackfan syndrome, n=30; other, n=22). Nineteen percent (19%) of patients were less than 16 years of age and 16% were greater than or equal to 65 years of age. There was a reduction in the absolute LIC from baseline to end of study (-4.2 mg Fe/g dry weight).
Study 3 (NCT00067080) was a multicenter, open-label, randomized trial of the safety and efficacy of deferasirox tablets for oral suspension relative to deferoxamine given for 1 year in patients with sickle cell disease and transfusional hemosiderosis. Patients were randomized to deferasirox tablets for oral suspension at doses of 5, 10, 20, or 30 mg per kg per day or subcutaneous deferoxamine at doses of 20-60 mg per kg per day for 5 days per week according to baseline LIC.
A total of 195 patients were treated in this study: 132 with deferasirox tablets for oral suspension and 63 with deferoxamine. Forty-four percent (44%) of patients were less than 16 years of age and 91% were black. At end of study, the mean change in LIC (as measured by magnetic susceptometry by a superconducting quantum interference device) in the per protocol-1 (PP-1) population, which consisted of patients who had at least 1 post-baseline LIC assessment, was -1.3 mg Fe/g dry weight for patients receiving deferasirox tablets for oral suspension (n=113) and -0.7 mg Fe/g dry weight for patients receiving deferoxamine (n=54).
One-hundred five (105) patients with thalassemia major and cardiac iron overload were enrolled in a study assessing the change in cardiac magnetic resonance imaging (MRI) T2* value (measured in milliseconds, [ms]) before and after treatment with deferasirox. Cardiac T2* values at baseline ranged from 5 to less than 20 ms. The geometric mean of cardiac T2* in the 68 patients who completed 3 years of deferasirox tablets for oral suspension therapy increased from 11.98 ms at baseline to 17.12 ms at 3 years. Cardiac T2* values improved in patients with severe cardiac iron overload (less than 10 ms) and in those with mild to moderate cardiac iron overload (greater than or equal to 10 to less than 20 ms). The clinical significance of these observations is unknown.
Six hundred twenty-seven (627) patients with MDS were enrolled across 5 uncontrolled trials. Two hundred thirty-nine of the 627 patients were enrolled in trials that limited enrollment to patients with IPSS Low or Intermediate 1 risk MDS, and the remaining 388 patients were enrolled in trials that did not specify MDS risk stratification but required a life expectancy of greater than 1 year. Planned duration of treatment in these trials ranged from 1 year (365 patients) to 5 years (47 patients). These trials evaluated the effects of deferasirox tablets for oral suspension therapy on parameters of iron overload, including LIC (125 patients) and serum ferritin (627 patients). The percent of patients completing planned duration of treatment was 51% in the largest 1-year study, 52% in the 3-year study and 22% in the 5-year study. The major causes for treatment discontinuation were withdrawal of consent, adverse reaction, and death. Over 1 year of follow-up across these pooled studies, mean change in serum ferritin was -332.8 (± 2615.59) mcg/L (n=593) and mean change in LIC was -5.9 (± 8.32) mg Fe/g dw (n=68). Results of these pooled studies in 627 patients with MDS suggest a progressive decrease in serum ferritin and LIC beyond 1 year in those patients who are able to continue deferasirox tablets for oral suspension.
Study 4 (TELESTO; NCT 00940602) was a randomized, double-blind, placebo-controlled trial performed in 225 patients with MDS (Low/Int-1 risk) and transfusional iron overload of which 149 were treated with deferasirox and 76 received placebo. The observed hazard ratio of 0.64 (95% CI: 0.42, 0.96) suggests a positive impact of deferasirox on event-free survival (EFS, a composite endpoint defined as death, worsening cardiac function, hospitalization for congestive heart failure, liver function impairment, liver cirrhosis, or progression to acute myeloid leukemia; whichever occurred first).
Non-Transfusion-Dependent Thalassemia
Study 5 (NCT00873041) was a randomized, double-blind, placebo-controlled trial of treatment with deferasirox tablets for oral suspension for patients 10 years of age or older with NTDT syndromes and iron overload. Eligible patients had an LIC of at least 5 mg Fe/g dw measured by R2 MRI and a serum ferritin exceeding 300 mcg/L at screening (2 consecutive values at least 14 days apart from each other). A total of 166 patients were randomized, 55 to the deferasirox tablets for oral suspension 5 mg/kg/day dose group, 55 to the deferasirox tablets for oral suspension 10 mg/kg/day dose group, and 56 to placebo (28 to each matching placebo group). Doses could be increased after 6 months if the LIC exceeded 7 mg Fe/g dw and the LIC reduction from baseline was less than 15%. The patients enrolled included 89 males and 77 females. The underlying disease was beta-thalassemia intermedia in 95 (57%) patients, HbE beta-thalassemia in 49 (30%) patients, and alpha-thalassemia in 22 (13%) patients. There were 17 pediatric patients in the study. Caucasians comprised 57% of the study population and Asians comprised 42%. The median baseline LIC (range) for all patients was 12.1 (2.6 to 49.1) mg Fe/g dw. Follow-up was for 1 year. The primary efficacy endpoint of change in LIC from baseline to Week 52 was statistically significant in favor of both deferasirox dose groups compared with placebo (p less than or equal to 0.001) (Table 5). Furthermore, a statistically significant dose effect of deferasirox was observed in favor of the 10 mg/kg/day dose group (10 versus 5 mg/kg/day, p=0.009). In a descriptive analysis, the target LIC (less than 5 mg Fe/g dw) was reached by 15 (27%) of 55 patients in the 10 mg/kg/day arm, 8 (15%) of 55 patients in the 5 mg/kg/day arm and 2 (4%) of 56 patients in the combined placebo groups.
Study 6 (NCT00873041) was an open-label trial of deferasirox tablets for oral suspension for the treatment of patients previously enrolled on Study 5, including cross-over to active treatment for those previously treated with placebo. The starting dose of deferasirox tablets for oral suspension in Study 6 was assigned based on the patient's LIC at completion of Study 5, being 20 mg/kg/day for an LIC exceeding 15 mg Fe/g dw, 10 mg/kg/day for LIC 3 to 15 mg Fe/g dw, and observation if the LIC was less than 3 mg Fe/g dw. Patients could continue on 5 mg/kg/day if they had previously exhibited at least a 30% reduction in LIC. Doses could be increased to a maximum of 20 mg/kg/day after 6 months if the LIC was more than 7 mg Fe/g dw and the LIC reduction from baseline was less than 15%. The primary efficacy endpoint in Study 6 was the proportion of patients achieving an LIC less than 5 mg Fe/g dw. A total of 133 patients were enrolled. Twenty patients began Study 6 with an LIC less than 5 mg Fe/g dw. Of the 113 patients with a baseline LIC of at least 5 mg Fe/g dw in Study 6, the target LIC (less than 5 mg Fe/g dw) was reached by 39 patients (35%). The responders included 4 (10%) of 39 patients treated at 20 mg/kg/day for a baseline LIC exceeding 15 mg Fe/g dw, and 31 (51%) of 61 patients treated at 10 mg/kg/day for a baseline LIC between 5 and 15 mg Fe/g dw. The absolute change in LIC at Week 52 by starting dose is shown in Table 5 below.
Study 7 (NCT01709838) was an open-label, single-arm, multi-center, 5-year study to evaluate the efficacy and safety of deferasirox tablets for oral suspension in iron overloaded patients with NTDT of 10 years of age or older. All patients started treatment on 10 mg/kg/day deferasirox tablets for oral suspension for four weeks. At Week 4, dose escalation was based on baseline LIC. At Week 24 and every 6 months thereafter, further dose adjustments were made according to the LIC at that visit. Treatment was interrupted when LIC <3 mg Fe/g dw or serum ferritin <300 ng/mL and was re-started at 10 mg/kg/day when LIC ≥5 mg Fe/g dw and serum ferritin ≥300 ng/mL. Throughout the study, the maximum dose of deferasirox tablets for oral suspension given was 30 mg/kg/day.
A total of 134 patients were enrolled in the study. Eligible patients were required to have an LIC of at least 5 mg Fe/g dw measured by R2 MRI and a serum ferritin at least of 300 ng/mL at screening. The mean absolute change of LIC from Baseline to Week 52 was -6.7 mg Fe/g dw. The reduction in LIC was sustained until Week 260 (5 years) with the mean absolute change in LIC from Baseline to Week 260 of -10.6 mg Fe/g dw. In the subset of patients with Baseline LIC >15 mg Fe/g dw (49 patients), 51.0% achieved a first LIC <5 mg Fe/g dw (95% CI: 37.5, 64.4) with a median time of 28.6 months. In the subset of patients with target LIC of <3 mg Fe/g dw (61 patients), 39.3% developed first LIC ≥5 mg Fe/g dw in the follow-up period, with a median time of 13.9 months.
Table 5. Absolute Change in LIC at Week 52 in Patients with NTDT:
| Deferasirox Tablets for Oral Suspension Starting Dosea | ||||
|---|---|---|---|---|
| Placebo | 5 mg/kg/day | 10 mg/kg/day | 20 mg/kg/day | |
| Study 5b | ||||
| Number of Patients | n=54 | n=51 | n=54 | - |
| Mean LIC at Baseline (mg Fe/g dw) | 16.1 | 13.4 | 14.4 | - |
| Mean Change (mg Fe/g dw) | +0.4 | -2.0 | -3.8 | - |
| (95% Confidence Interval) | (-0.6, +1.3) | (-2.9, -1.0) | (-4.8, -2.9) | - |
| Study 6 | ||||
| Number of Patients | - | n=8 | n=77 | n=43 |
| Mean LIC at Baseline (mg Fe/g dw) | - | 5.6 | 8.8 | 23.5 |
| Mean Change (mg Fe/g dw) | - | -1.5 | -2.8 | -9.1 |
| (95% Confidence Interval) | - | (-3.7, +0.7) | (-3.4, -2.2) | (-11.0, -7.3) |
| Study 7 | ||||
| Number of Patients | - | - | n=127 | - |
| Mean LIC at Baseline (mg Fe/g dw) | - | - | 15.1 | - |
| Mean Change (mg Fe/g dw) | - | - | -6.7 | - |
| (95% Confidence Interval) | - | - | (-7.9, -5.5) | - |
Abbreviation: LIC, liver iron concentration; NTDT, non-transfusion-dependent thalassemia.
a Randomized dose in Study 5 or assigned starting dose in Study 6 and Study 7.
b Least square mean change for Study 5.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.