JANUMET Film-coated tablet Ref.[10494] Active ingredients: Metformin Metformin and Sitagliptin Sitagliptin
Source: European Medicines Agency (EU) Revision Year: 2023 Publisher: Merck Sharp & Dohme B.V., Waarderweg 39, 2031 BN Haarlem, The Netherlands
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Drugs used in diabetes, Combinations of oral blood glucose lowering drugs
ATC code: A10BD07
Janumet combines two antihyperglycaemic medicinal products with complementary mechanisms of action to improve glycaemic control in patients with type 2 diabetes: sitagliptin phosphate, a dipeptidyl peptidase 4 (DPP-4) inhibitor, and metformin hydrochloride, a member of the biguanide class.
Sitagliptin
Mechanism of action
Sitagliptin phosphate is an orally-active, potent, and highly selective inhibitor of the dipeptidyl peptidase 4 (DPP-4) enzyme for the treatment of type 2 diabetes. The DPP-4 inhibitors are a class of agents that act as incretin enhancers. By inhibiting the DPP-4 enzyme, sitagliptin increases the levels of two known active incretin hormones, glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP). The incretins are part of an endogenous system involved in the physiologic regulation of glucose homeostasis. When blood glucose concentrations are normal or elevated, GLP-1 and GIP increase insulin synthesis and release from pancreatic beta cells. GLP-1 also lowers glucagon secretion from pancreatic alpha cells, leading to reduced hepatic glucose production. When blood glucose levels are low, insulin release is not enhanced and glucagon secretion is not suppressed. Sitagliptin is a potent and highly selective inhibitor of the enzyme DPP-4 and does not inhibit the closely-related enzymes DPP-8 or DPP-9 at therapeutic concentrations. Sitagliptin differs in chemical structure and pharmacological action from GLP-1 analogues, insulin, sulphonylureas or meglitinides, biguanides, peroxisome proliferator-activated receptor gamma (PPAR) agonists, alpha-glucosidase inhibitors, and amylin analogues.
In a two-day study in healthy subjects, sitagliptin alone increased active GLP-1 concentrations, whereas metformin alone increased active and total GLP-1 concentrations to similar extents. Co-administration of sitagliptin and metformin had an additive effect on active GLP-1 concentrations. Sitagliptin, but not metformin, increased active GIP concentrations.
Clinical efficacy and safety
Overall, sitagliptin improved glycaemic control when used as monotherapy or in combination treatment in adult patients with type 2 diabetes.
In clinical trials, sitagliptin as monotherapy improved glycaemic control with significant reductions in haemoglobin A1c (HbA1c) and fasting and postprandial glucose. Reduction in fasting plasma glucose (FPG) was observed at 3 weeks, the first time point at which FPG was measured. The observed incidence of hypoglycaemia in patients treated with sitagliptin was similar to placebo. Body weight did not increase from baseline with sitagliptin therapy. Improvements in surrogate markers of beta cell function, including HOMA-β (Homeostasis Model Assessment-β), proinsulin to insulin ratio, and measures of beta cell responsiveness from the frequently-sampled meal tolerance test were observed.
Studies of sitagliptin in combination with metformin
In a 24-week, placebo-controlled clinical study to evaluate the efficacy and safety of the addition of sitagliptin 100 mg once daily to ongoing metformin, sitagliptin provided significant improvements in glycaemic parameters compared with placebo. Change from baseline in body weight was similar for patients treated with sitagliptin relative to placebo. In this study there was a similar incidence of hypoglycaemia reported for patients treated with sitagliptin or placebo.
In a 24-week placebo-controlled factorial study of initial therapy, sitagliptin 50 mg twice daily in combination with metformin (500 mg or 1,000 mg twice daily) provided significant improvements in glycaemic parameters compared with either monotherapy. The decrease in body weight with the combination of sitagliptin and metformin was similar to that observed with metformin alone or placebo; there was no change from baseline for patients on sitagliptin alone. The incidence of hypoglycaemia was similar across treatment groups.
Study of sitagliptin in combination with metformin and a sulphonylurea
A 24-week placebo-controlled study was designed to evaluate the efficacy and safety of sitagliptin (100 mg once daily) added to glimepiride (alone or in combination with metformin). The addition of sitagliptin to glimepiride and metformin provided significant improvements in glycaemic parameters. Patients treated with sitagliptin had a modest increase in body weight (+1.1 kg) compared to those given placebo.
Study of sitagliptin in combination with metformin and a PPARγ agonist
A 26-week placebo-controlled study was designed to evaluate the efficacy and safety of sitagliptin (100 mg once daily) added to the combination of pioglitazone and metformin. The addition of sitagliptin to pioglitazone and metformin provided significant improvements in glycaemic parameters. Change from baseline in body weight was similar for patients treated with sitagliptin relative to placebo. The incidence of hypoglycaemia was also similar in patients treated with sitagliptin or placebo.
Study of sitagliptin in combination with metformin and insulin
A 24-week placebo-controlled study was designed to evaluate the efficacy and safety of sitagliptin (100 mg once daily) added to insulin (at a stable dose for at least 10 weeks) with or without metformin (at least 1,500 mg). In patients taking pre-mixed insulin, the mean daily dose was 70.9 U/day. In patients taking non-pre-mixed (intermediate/long-acting) insulin, the mean daily dose was 44.3 U/day. Data from the 73% of patients who were taking metformin are presented in Table 2. The addition of sitagliptin to insulin provided significant improvements in glycaemic parameters. There was no meaningful change from baseline in body weight in either group.
Table 2. HbA1c results in placebo-controlled combination therapy studies of sitagliptin and metformin*:
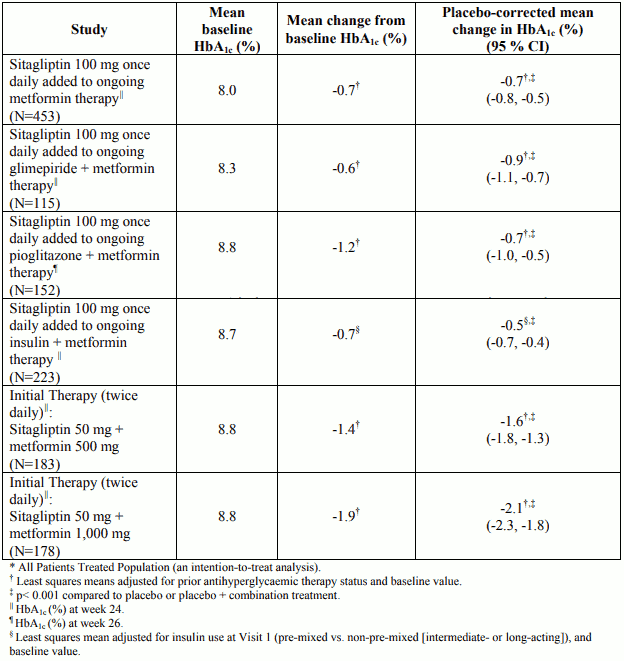
In a 52-week study, comparing the efficacy and safety of the addition of sitagliptin 100 mg once daily or glipizide (a sulphonylurea) in patients with inadequate glycaemic control on metformin monotherapy, sitagliptin was similar to glipizide in reducing HbA1c (-0.7% mean change from baselines at week 52, with baseline HbA1c of approximately 7.5% in both groups). The mean glipizide dose used in the comparator group was 10 mg per day with approximately 40% of patients requiring a glipizide dose of ≤5 mg/day throughout the study. However, more patients in the sitagliptin group discontinued due to lack of efficacy than in the glipizide group. Patients treated with sitagliptin exhibited a significant mean decrease from baseline in body weight (-1.5 kg) compared to a significant weight gain in patients administered glipizide (+1.1 kg). In this study, the proinsulin to insulin ratio, a marker of efficiency of insulin synthesis and release, improved with sitagliptin and deteriorated with glipizide treatment. The incidence of hypoglycaemia in the sitagliptin group (4.9%) was significantly lower than that in the glipizide group (32.0%).
A 24-week placebo-controlled study involving 660 patients was designed to evaluate the insulin-sparing efficacy and safety of sitagliptin (100 mg once daily) added to insulin glargine with or without metformin (at least 1,500 mg) during intensification of insulin therapy. Among patients taking metformin, baseline HbA1c was 8.70% and baseline insulin dose was 37 IU/day. Patients were instructed to titrate their insulin glargine dose based on fingerstick fasting glucose values. Among patients taking metformin, at Week 24, the increase in daily insulin dose was 19 IU/day in patients treated with sitagliptin and 24 IU/day in patients treated with placebo. The reduction in HbA1c for patients treated with sitagliptin, metformin, and insulin was -1.35% compared to -0.90% for patients treated with placebo, metformin, and insulin, a difference of -0.45% [95% CI: -0.62, -0.29]. The incidence of hypoglycaemia was 24.9% for patients treated with sitagliptin, metformin, and insulin and 37.8% for patients treated with placebo, metformin, and insulin. The difference was mainly due to a higher percentage of patients in the placebo group experiencing 3 or more episodes of hypoglycaemia (9.1 vs. 19.8%). There was no difference in the incidence of severe hypoglycaemia.
Metformin
Mechanism of action
Metformin is a biguanide with antihyperglycaemic effects, lowering both basal and postprandial plasma glucose. It does not stimulate insulin secretion and therefore does not produce hypoglycaemia.
Metformin may act via three mechanisms:
- by reduction of hepatic glucose production by inhibiting gluconeogenesis and glycogenolysis
- in muscle, by modestly increasing insulin sensitivity, improving peripheral glucose uptake and utilisation
- by delaying intestinal glucose absorption.
Metformin stimulates intracellular glycogen synthesis by acting on glycogen synthase. Metformin increases the transport capacity of specific types of membrane glucose transporters (GLUT-1 and GLUT-4).
Clinical efficacy and safety
In humans, independently of its action on glycaemia, metformin has favourable effects on lipid metabolism. This has been shown at therapeutic doses in controlled, medium-term or long-term clinical studies: metformin reduces total cholesterol, LDLc and triglyceride levels.
The prospective randomised (UKPDS) study has established the long-term benefit of intensive blood glucose control in type 2 diabetes. Analysis of the results for overweight patients treated with metformin after failure of diet alone showed:
- a significant reduction of the absolute risk of any diabetes-related complication in the metformin group (29.8 events/1,000 patient-years) versus diet alone (43.3 events/1,000 patient-years), p=0.0023, and versus the combined sulphonylurea and insulin monotherapy groups (40.1 events/1,000 patient-years), p=0.0034
- a significant reduction of the absolute risk of any diabetes-related mortality: metformin 7.5 events/1,000 patient-years, diet alone 12.7 events/1,000 patient-years, p=0.017
- a significant reduction of the absolute risk of overall mortality: metformin 13.5 events/1,000 patient-years versus diet alone 20.6 events/1,000 patient-years, (p=0.011), and versus the combined sulphonylurea and insulin monotherapy groups 18.9 events/1,000 patient-years (p=0.021)
- a significant reduction in the absolute risk of myocardial infarction: metformin 11 events/1,000 patient-years, diet alone 18 events/1,000 patient-years, (p=0.01).
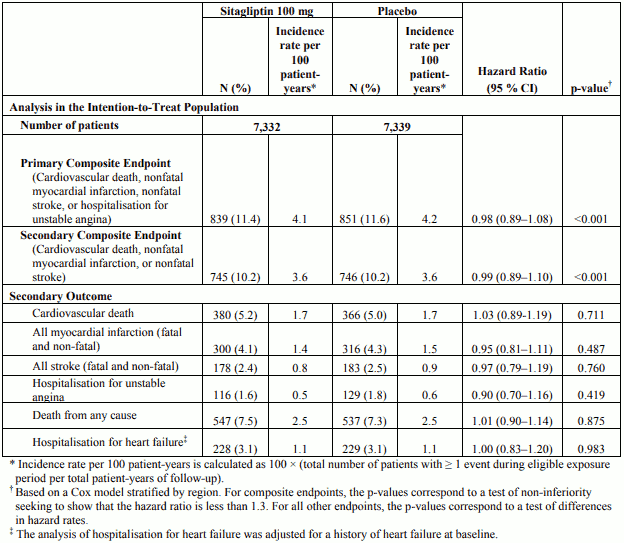
The TECOS was a randomised study in 14,671 patients in the intention-to-treat population with an HbA1c of ≥6.5 to 8.0% with established CV disease who received sitagliptin (7,332) 100 mg daily (or 50 mg daily if the baseline eGFR was ≥30 and <50 mL/min/1.73 m²) or placebo (7,339) added to usual care targeting regional standards for HbA1c and CV risk factors. Patients with an eGFR <30 mL/min/1.73 m² were not to be enrolled in the study. The study population included 2,004 patients ≥75 years of age and 3,324 patients with renal impairment (eGFR <60 mL/min/1.73 m²).
Over the course of the study, the overall estimated mean (SD) difference in HbA1c between the sitagliptin and placebo groups was 0.29% (0.01), 95% CI (-0.32, -0.27); p <0.001.
The primary cardiovascular endpoint was a composite of the first occurrence of cardiovascular death, nonfatal myocardial infarction, nonfatal stroke, or hospitalisation for unstable angina. Secondary cardiovascular endpoints included the first occurrence of cardiovascular death, nonfatal myocardial infarction, or nonfatal stroke; first occurrence of the individual components of the primary composite; all-cause mortality; and hospital admissions for congestive heart failure.
After a median follow up of 3 years, sitagliptin, when added to usual care, did not increase the risk of major adverse cardiovascular events or the risk of hospitalisation for heart failure compared to usual care without sitagliptin in patients with type 2 diabetes (Table 3).
Table 3. Rates of Composite Cardiovascular Outcomes and Key Secondary Outcomes:
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with Janumet in all subsets of the paediatric population in type 2 diabetes mellitus (see section 4.2 for information on paediatric use).
The safety and efficacy of the addition of sitagliptin in paediatric patients aged 10 to 17 years with type 2 diabetes and inadequate glycaemic control on metformin with or without insulin was assessed in two studies over 54 weeks. The addition of sitagliptin (administered as sitagliptin + metformin or sitagliptin + metformin extended release (XR)) was compared to the addition of placebo to metformin or metformin XR.
While superiority of HbA1c reduction was demonstrated for sitagliptin + metformin/sitagliptin + metformin XR over metformin at Week 20 in the pooled analysis of these two studies, results from the individual studies were inconsistent. Furthermore, greater efficacy for sitagliptin + metformin/sitagliptin + metformin XR compared to metformin was not observed at Week 54. Therefore, Janumet should not be used in paediatric patients aged 10 to 17 years old because of insufficient efficacy (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Janumet
A bioequivalence study in healthy subjects demonstrated that the Janumet (sitagliptin/metformin hydrochloride) combination tablets are bioequivalent to co-administration of sitagliptin phosphate and metformin hydrochloride as individual tablets.
The following statements reflect the pharmacokinetic properties of the individual active substances of Janumet.
Sitagliptin
Absorption
Following oral administration of a 100-mg dose to healthy subjects, sitagliptin was rapidly absorbed, with peak plasma concentrations (median Tmax) occurring 1 to 4 hours post-dose, mean plasma AUC of sitagliptin was 8.52 M•hr, Cmax was 950 nM. The absolute bioavailability of sitagliptin is approximately 87%. Since co-administration of a high-fat meal with sitagliptin had no effect on the pharmacokinetics, sitagliptin may be administered with or without food.
Plasma AUC of sitagliptin increased in a dose-proportional manner. Dose-proportionality was not established for Cmax and C24hr (Cmax increased in a greater than dose-proportional manner and Csub>24hr increased in a less than dose-proportional manner).
Distribution
The mean volume of distribution at steady state following a single 100-mg intravenous dose of sitagliptin to healthy subjects is approximately 198 litres. The fraction of sitagliptin reversibly bound to plasma proteins is low (38%).
Biotransformation
Sitagliptin is primarily eliminated unchanged in urine, and metabolism is a minor pathway. Approximately 79% of sitagliptin is excreted unchanged in the urine.
Following a [14C]sitagliptin oral dose, approximately 16% of the radioactivity was excreted as metabolites of sitagliptin. Six metabolites were detected at trace levels and are not expected to contribute to the plasma DPP-4 inhibitory activity of sitagliptin. In vitro studies indicated that the primary enzyme responsible for the limited metabolism of sitagliptin was CYP3A4, with contribution from CYP2C8.
In vitro data showed that sitagliptin is not an inhibitor of CYP isoenzymes CYP3A4, 2C8, 2C9, 2D6, 1A2, 2C19 or 2B6, and is not an inducer of CYP3A4 and CYP1A2.
Elimination
Following administration of an oral [14C]sitagliptin dose to healthy subjects, approximately 100% of the administered radioactivity was eliminated in faeces (13%) or urine (87%) within one week of dosing. The apparent terminal t½ following a 100-mg oral dose of sitagliptin was approximately 12.4 hours. Sitagliptin accumulates only minimally with multiple doses. The renal clearance was approximately 350 mL/min.
Elimination of sitagliptin occurs primarily via renal excretion and involves active tubular secretion. Sitagliptin is a substrate for human organic anion transporter-3 (hOAT-3), which may be involved in the renal elimination of sitagliptin. The clinical relevance of hOAT-3 in sitagliptin transport has not been established. Sitagliptin is also a substrate of p-glycoprotein, which may also be involved in mediating the renal elimination of sitagliptin. However, ciclosporin, a p-glycoprotein inhibitor, did not reduce the renal clearance of sitagliptin. Sitagliptin is not a substrate for OCT2 or OAT1 or PEPT1/2 transporters. In vitro, sitagliptin did not inhibit OAT3 (IC50=160 M) or p-glycoprotein (up to 250 M) mediated transport at therapeutically relevant plasma concentrations. In a clinical study sitagliptin had a small effect on plasma digoxin concentrations indicating that sitagliptin may be a mild inhibitor of p-glycoprotein.
Characteristics in patients
The pharmacokinetics of sitagliptin were generally similar in healthy subjects and in patients with type 2 diabetes.
Renal impairment
A single-dose, open-label study was conducted to evaluate the pharmacokinetics of a reduced dose of sitagliptin (50 mg) in patients with varying degrees of chronic renal impairment compared to normal healthy control subjects. The study included patients with mild, moderate, and severe renal impairment, as well as patients with ESRD on haemodialysis. In addition, the effects of renal impairment on sitagliptin pharmacokinetics in patients with type 2 diabetes and mild, moderate, or severe renal impairment (including ESRD) were assessed using population pharmacokinetic analyses.
Compared to normal healthy control subjects, plasma AUC of sitagliptin was increased by approximately 1.2-fold and 1.6-fold in patients with mild renal impairment (GFR ≥60 to <90 mL/min) and patients with moderate renal impairment (GFR ≥45 to <60 mL/min), respectively. Because increases of this magnitude are not clinically relevant, dosage adjustment in these patients is not necessary.
Plasma AUC of sitagliptin was increased approximately 2-fold in patients with moderate renal impairment (GFR ≥30 to <45 mL/min), and approximately 4-fold in patients with severe renal impairment (GFR <30 mL/min), including patients with ESRD on haemodialysis. Sitagliptin was modestly removed by haemodialysis (13.5% over a 3- to 4-hour haemodialysis session starting 4 hours post-dose).
Hepatic impairment
No dose adjustment for sitagliptin is necessary for patients with mild or moderate hepatic impairment (Child-Pugh score 9). There is no clinical experience in patients with severe hepatic impairment (Child-Pugh score >9). However, because sitagliptin is primarily renally eliminated, severe hepatic impairment is not expected to affect the pharmacokinetics of sitagliptin.
Elderly
No dose adjustment is required based on age. Age did not have a clinically meaningful impact on the pharmacokinetics of sitagliptin based on a population pharmacokinetic analysis of Phase I and Phase II data. Elderly subjects (65 to 80 years) had approximately 19% higher plasma concentrations of sitagliptin compared to younger subjects.
Paediatric population
The pharmacokinetics of sitagliptin (single dose of 50 mg, 100 mg or 200 mg) were investigated in paediatric patients (10 to 17 years of age) with type 2 diabetes. In this population, the dose adjusted AUC of sitagliptin in plasma was approximately 18% lower compared to adult patients with type 2 diabetes for a 100 mg dose. No studies with sitagliptin have been performed in paediatric patients <10 years of age.
Other patient characteristics
No dose adjustment is necessary based on gender, race, or body mass index (BMI). These characteristics had no clinically meaningful effect on the pharmacokinetics of sitagliptin based on a composite analysis of Phase I pharmacokinetic data and on a population pharmacokinetic analysis of Phase I and Phase II data.
Metformin
Absorption
After an oral dose of metformin, Tmax is reached in 2.5 h. Absolute bioavailability of a 500 mg metformin tablet is approximately 50-60% in healthy subjects. After an oral dose, the non-absorbed fraction recovered in faeces was 20-30%.
After oral administration, metformin absorption is saturable and incomplete. It is assumed that the pharmacokinetics of metformin absorption is non-linear. At the usual metformin doses and dosing schedules, steady state plasma concentrations are reached within 24-48 h and are generally less than 1 µg/mL. In controlled clinical trials, maximum metformin plasma levels (Cmax) did not exceed 5 µg/mL, even at maximum doses.
Food decreases the extent and slightly delays the absorption of metformin. Following administration of a dose of 850 mg, a 40% lower plasma peak concentration, a 25% decrease in AUC and a 35 min prolongation of time to peak plasma concentration was observed. The clinical relevance of this decrease is unknown.
Distribution
Plasma protein binding is negligible. Metformin partitions into erythrocytes. The blood peak is lower than the plasma peak and appears at approximately the same time. The red blood cells most likely represent a secondary compartment of distribution. The mean Vd ranged between 63-276 L.
Biotransformation
Metformin is excreted unchanged in the urine. No metabolites have been identified in humans.
Elimination
Renal clearance of metformin is >400 mL/min, indicating that metformin is eliminated by glomerular filtration and tubular secretion. Following an oral dose, the apparent terminal elimination half-life is approximately 6.5 h. When renal function is impaired, renal clearance is decreased in proportion to that of creatinine and thus the elimination half-life is prolonged, leading to increased levels of metformin in plasma.
5.3. Preclinical safety data
No animal studies have been conducted with Janumet.
In 16-week studies in which dogs were treated with either metformin alone or a combination of metformin and sitagliptin, no additional toxicity was observed from the combination. The NOEL in these studies was observed at exposures to sitagliptin of approximately 6 times the human exposure and to metformin of approximately 2.5 times the human exposure. The following data are findings in studies performed with sitagliptin or metformin individually.
Sitagliptin
Renal and liver toxicity were observed in rodents at systemic exposure values 58 times the human exposure level, while the no-effect level was found at 19 times the human exposure level. Incisor teeth abnormalities were observed in rats at exposure levels 67 times the clinical exposure level; the no-effect level for this finding was 58-fold based on the 14-week rat study. The relevance of these findings for humans is unknown. Transient treatment-related physical signs, some of which suggest neural toxicity, such as open-mouth breathing, salivation, white foamy emesis, ataxia, trembling, decreased activity, and/or hunched posture were observed in dogs at exposure levels approximately 23 times the clinical exposure level. In addition, very slight to slight skeletal muscle degeneration was also observed histologically at doses resulting in systemic exposure levels of approximately 23 times the human exposure level. A no-effect level for these findings was found at an exposure 6-fold the clinical exposure level.
Sitagliptin has not been demonstrated to be genotoxic in preclinical studies. Sitagliptin was not carcinogenic in mice. In rats, there was an increased incidence of hepatic adenomas and carcinomas at systemic exposure levels 58 times the human exposure level. Since hepatotoxicity has been shown to correlate with induction of hepatic neoplasia in rats, this increased incidence of hepatic tumours in rats was likely secondary to chronic hepatic toxicity at this high dose. Because of the high safety margin (19-fold at this no-effect level), these neoplastic changes are not considered relevant for the situation in humans.
No treatment related effects on fertility were observed in male and female rats given sitagliptin prior to and throughout mating.
In a pre-/post-natal development study performed in rats sitagliptin showed no adverse effects.
Reproductive toxicity studies showed a slight treatment-related increased incidence of foetal rib malformations (absent, hypoplastic and wavy ribs) in the offspring of rats at systemic exposure levels more than 29 times the human exposure levels. Maternal toxicity was seen in rabbits at more than 29 times the human exposure levels. Because of the high safety margins, these findings do not suggest a relevant risk for human reproduction. Sitagliptin is secreted in considerable amounts into the milk of lactating rats (milk/plasma ratio: 4:1).
Metformin
Preclinical data for metformin reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential, toxicity to reproduction.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.