JEMPERLI Concentrate for solution for infusion Ref.[27949] Active ingredients: Dostarlimab
Source: European Medicines Agency (EU) Revision Year: 2025 Publisher: GlaxoSmithKline (Ireland) Limited, 12 Riverwalk, Citywest Business Campus, Dublin 24, Ireland
4.1. Therapeutic indications
JEMPERLI is indicated in combination with carboplatin and paclitaxel for the first-line treatment of adult patients with primary advanced or recurrent endometrial cancer (EC) and who are candidates for systemic therapy.
JEMPERLI is indicated as monotherapy for the treatment of adult patients with mismatch repair deficient (dMMR)/microsatellite instability-high (MSI-H) recurrent or advanced EC that has progressed on or following prior treatment with a platinum-containing regimen.
4.2. Posology and method of administration
Therapy must be initiated and supervised by specialist physicians experienced in the treatment of cancer.
The identification of dMMR/MSI-H tumour status should be determined using a validated testing method such as IHC, PCR or NGS* (see section 5.1 for information on assays used in the studies).
* IHC = immunohistochemistry; PCR = polymerase chain reaction; NGS = next-generation sequencing.
Posology
JEMPERLI in combination with carboplatin and paclitaxel
When JEMPERLI is administered in combination with carboplatin and paclitaxel, refer to the full Prescribing Information for the combination products (see also section 5.1).
The recommended dose is 500 mg dostarlimab every 3 weeks in combination with carboplatin and paclitaxel every 3 weeks for 6 cycles followed by 1000 mg dostarlimab as monotherapy every 6 weeks for all cycles thereafter.
The dosage regimen in combination with carboplatin and paclitaxel is presented in Table 1.
Table 1. Dosage regimen for JEMPERLI in combination with carboplatin and paclitaxel:
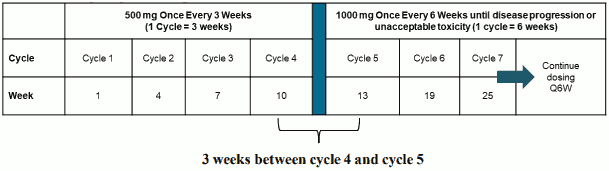
a Administer dostarlimab prior to carboplatin and paclitaxel on the same day.
Administration of dostarlimab should continue according to the recommended schedule until disease progression or unacceptable toxicity, or for a duration of up to 3 years (see section 5.1).
JEMPERLI monotherapy
The recommended dose as monotherapy is 500 mg dostarlimab every 3 weeks for 4 cycles followed by 1000 mg every 6 weeks for all cycles thereafter.
The dosage regimen as monotherapy is presented in Table 2.
Table 2. Dosage regimen for JEMPERLI as monotherapy:

Administration of dostarlimab should continue according to the recommended schedule until disease progression or unacceptable toxicity (see section 5.1).
Dose modifications
Dose reduction is not recommended. Dosing delay or discontinuation may be required based on individual safety and tolerability. Recommended modifications to manage adverse reactions are provided in Table 3.
Detailed guidelines for the management of immune-related adverse reactions and infusion-related reactions are described in section 4.4.
Table 3. Recommended dose modifications for JEMPERLI:
| Immune-related adverse reactions | Severity gradea | Dose modification,/b> |
| Colitis | 2 or 3 | Withhold dose. Restart dosing when toxicity resolves to grade 0 or 1. |
| 4 | Permanently discontinue. | |
| Hepatitis | Grade 2 with ASTb or ALTc > 3 and up to 5 × ULNd or total bilirubin > 1.5 and up to 3 × ULN | Withhold dose. Restart dosing when toxicity resolves to grade 0 or 1. |
| Grade ≥ 3 with AST or ALT > 5 × ULN or total bilirubin > 3 × ULN | Permanently discontinue (see exception below)e. | |
| Type 1 diabetes mellitus (T1DM) | 3 or 4 (hyperglycaemia) | Withhold dose. Restart dosing in appropriately managed, clinically and metabolically stable patients. |
| Hypophysitis or adrenal insufficiency | 2, 3 or 4 | Withhold dose. Restart dosing when toxicity resolves to grade 0 or 1. Permanently discontinue for recurrence or worsening while on adequate hormonal therapy. |
| Hypothyroidism or hyperthyroidism | 3 or 4 | Withhold dose. Restart dosing when toxicity resolves to grade 0 or 1. |
| Pneumonitis | 2 | Withhold dose. Restart dosing when toxicity resolves to grade 0 or 1. If grade 2 recurs, permanently discontinue. |
| 3 or 4 | Permanently discontinue. | |
| Nephritis | 2 | Withhold dose. Restart dosing when toxicity resolves to grade 0 or 1. |
| 3 or 4 | Permanently discontinue. | |
| Exfoliative dermatologic conditions (e.g. SJSf, TENg, DRESSh) | Suspected | Withhold dose for any grade. Restart dosing if not confirmed and when toxicity resolves to grade 0 or 1. |
| Confirmed | Permanently discontinue. | |
| Myocarditis | 2, 3 or 4 | Permanently discontinue. |
| Severe neurological toxicities (myasthenic syndrome/myasthenia gravis, Guillain-Barré syndrome, encephalitis, transverse myelitis) | 2, 3 or 4 | Permanently discontinue. |
| Other immune-related adverse reactions (including but not limited to myositis, sarcoidosis, autoimmune haemolytic anaemia, pancreatitis, iridocyclitis, uveitis, diabetic ketoacidosis, arthralgia, solid organ transplant rejection, graft- versus-host disease) | 3 | Withhold dose. Restart dosing when toxicity resolves to grade 0 or 1. |
| 4 | Permanently discontinue. | |
| Recurrence of immune-related adverse reactions after resolution to ≤ grade 1 (except for pneumonitis, see above) | 3 or 4 | Permanently discontinue. |
| Other adverse reactions | Severity gradea | Dose modification |
| Infusion-related reactions | 2 | Withhold dose. If resolved within 1 hour of stopping, may be restarted at 50% of the original infusion rate, or restart when symptoms resolve with pre-medication. If grade 2 recurs with adequate premedication, permanently discontinue. |
| 3 or 4 | Permanently discontinue. |
a Toxicity graded per National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 5.0.
b AST = aspartate aminotransferase
c ALT = alanine aminotransferase
d ULN = upper limit of normal
e For patients with liver metastases who begin treatment with grade 2 increase of AST or ALT, if AST or ALT increases by ≥50% relative to baseline and lasts for at least 1 week, then treatment should be discontinued.
f SJS = Stevens-Johnson syndrome
g TEN = toxic epidermal necrolysis
h DRESS = drug reaction with eosinophilia and systemic symptoms.
Patient Card
All prescribers of JEMPERLI should inform patients about the Patient Card, explaining what to do should they experience any symptom of immune-related adverse reactions. The physician will provide the Patient Card to each patient.
Special populations
Elderly
No dose adjustment is recommended for patients who are aged 65 years or over.
There are limited clinical data with dostarlimab in patients aged 75 years or over (see section 5.1).
Renal impairment
No dose adjustment is recommended for patients with mild or moderate renal impairment. There are limited data in patients with severe renal impairment or end-stage renal disease undergoing dialysis (see section 5.2).
Hepatic impairment
No dose adjustment is recommended for patients with mild hepatic impairment. There are limited data in patients with moderate hepatic impairment and no data in patients with severe hepatic impairment (see section 5.2).
Paediatric population
The safety and efficacy of JEMPERLI in children and adolescents aged under 18 years have not been established. No data are available.
Method of administration
JEMPERLI is for intravenous infusion only. JEMPERLI should be administered by intravenous infusion using an intravenous infusion pump over 30 minutes.
JEMPERLI must not be administered as an intravenous push or bolus injection.
For instructions on dilution of the medicinal product before administration, see section 6.6.
4.9. Overdose
If overdose is suspected, the patient should be monitored for any signs or symptoms of adverse reactions or effects, and appropriate symptomatic treatment instituted.
6.3. Shelf life
Unopened vial: 3 years.
After dilution: If not used immediately, chemical and physical in-use stability has been demonstrated for 24 hours at 2°C-8°C and 6 hours at room temperature (up to 25°C) from the time of preparation/dilution until the end of administration.
6.4. Special precautions for storage
Store in a refrigerator 2°C-8°C.
Do not freeze.
Store in the original package in order to protect from light.
For storage conditions after dilution of the medicinal product, see section 6.3.
6.5. Nature and contents of container
10 mL type I borosilicate clear glass vial, with a grey chlorobutyl elastomer stopper laminated with fluoropolymer, sealed with an aluminium flip-off cap containing 500 mg dostarlimab.
Each carton contains one vial.
6.6. Special precautions for disposal and other handling
Preparation/dilution
Parenteral medicinal products should be inspected visually for particulate matter and discolouration prior to administration. JEMPERLI is a slightly opalescent colourless to yellow solution. Discard the vial if visible particles are observed.
JEMPERLI is compatible with an IV bag made of polyvinyl chloride (PVC) with or without di(2-ethylhexyl) phthalate (DEHP), ethylene vinyl acetate, polyethylene (PE), polypropylene (PP) or polyolefin blend (PP+PE), and a syringe made from PP.
For the 500 mg dose, withdraw 10 mL of JEMPERLI from a vial and transfer into an intravenous bag containing sodium chloride 9 mg/mL (0.9%) solution for injection, or glucose 50 mg/mL (5%) solution for injection. The final concentration of the diluted solution should be between 2 mg/mL and 10 mg/mL. The total volume of the infusion solution must not exceed 250 mL. This may require withdrawing a volume of diluent from the intravenous bag prior to adding a volume of JEMPERLI into the IV bag.
- For example, if preparing a 500 mg dose in a 250 mL diluent intravenous bag, to achieve a 2 mg/mL concentration would require withdrawing 10 mL of diluent from the 250 mL intravenous bag. Then, 10 mL of JEMPERLI would be withdrawn from the vial and transferred into the intravenous bag.
For the 1000 mg dose, withdraw 10 mL of JEMPERLI from each of two vials (withdraw 20 mL total) and transfer into an intravenous bag containing sodium chloride 9 mg/mL (0.9%) solution for injection, or glucose 50 mg/mL (5%) solution for injection. The final concentration of the diluted solution should be between 4 mg/mL and 10 mg/mL. The total volume of the infusion solution must not exceed 250 mL. This may require withdrawing a volume of diluent from the IV bag prior to adding a volume of JEMPERLI into the intravenous bag.
- For example, if preparing a 1000 mg dose in a 250 mL diluent intravenous bag, to achieve a 4 mg/mL concentration would require withdrawing 20 mL of diluent from the 250 mL intravenous bag. Then, 10 mL of JEMPERLI would be withdrawn from each of two vials, totaling 20 mL, and transferred into the intravenous bag.
Mix diluted solution by gentle inversion. Do not shake the final infusion bag. Discard any unused portion left in the vial.
Storage
Store in the original carton until time of preparation in order to protect from light. The prepared dose may be stored either:
- At room temperature up to 25ºC for no more than 6 hours from the time of dilution until the end of infusion.
- Under refrigeration at 2°C to 8°C for no more than 24 hours from time of dilution until end of infusion. If refrigerated, allow the diluted solution to come to room temperature prior to administration.
Administration
JEMPERLI should be administered by intravenous infusion using an intravenous infusion pump over 30 minutes by a health care practitioner. Tubing should be made of PVC, platinum cured silicon or PP; fittings made from PVC or polycarbonate and needles made from stainless steel. A 0.2 or 0.22 micron inline polyethersulfone (PES) filter must be used during administration of JEMPERLI.
JEMPERLI must not be administered as an intravenous push or bolus injection.
Do not co-administer other medicinal products through the same infusion line.
Any unused medicinal product or waste material should be disposed of in accordance with local requirements.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.