KESIMPTA Solution for injection Ref.[27863] Active ingredients: Ofatumumab
Source: European Medicines Agency (EU) Revision Year: 2026 Publisher: Novartis Europharm Limited, Vista Building, Elm Park, Merrion Road, Dublin 4, Ireland
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: immunosuppressants, monoclonal antibodies
ATC code: L04AG12
Mechanism of action
Ofatumumab is a fully human anti-CD20 monoclonal immunoglobulin G1 (IgG1) antibody. The CD20 molecule is a transmembrane phosphoprotein expressed on B lymphocytes from the pre-B to mature B lymphocyte stage. The CD20 molecule is also expressed on a small fraction of activated T cells. A subcutaneous route of administration of ofatumumab and subsequent release/absorption from the tissue allows a gradual interaction with B cells.
The binding of ofatumumab to CD20 induces lysis of CD20+ B cells primarily through complement-dependent cytotoxicity (CDC) and, to a lesser extent, through antibody-dependent cell-mediated cytotoxicity (ADCC). Ofatumumab has also been shown to induce cell lysis in both high and low CD20 expressing cells. CD20-expressing T cells are also depleted by ofatumumab.
Pharmacodynamic effects
B-cell depletion
In the RMS clinical studies using ofatumumab 20 mg every 4 weeks, after an initial dose regimen of 20 mg on days 1, 7, and 14, administration resulted in a rapid and sustained reduction of B cells to below LLN (defined as 40 cells/µl) as early as two weeks after treatment initiation. Before initiation of the maintenance phase starting at week 4, total B-cell levels of <10 cells/µl were reached in 94% of patients, increasing to 98% of patients at week 12, and were sustained for as long as 120 weeks (i.e. while on study treatment).
B-cell repletion
Data from RMS phase III clinical studies indicate a median time to B-cell recovery to LLN or baseline value of 24.6 weeks post treatment discontinuation. PK-B cell modelling and simulation for B-cell repletion corroborate this data, predicting median time to B-cell recovery to LLN of 23 weeks post treatment discontinuation.
Immunogenicity
In RMS phase III studies, the overall incidence of treatment-induced anti-drug antibodies (ADAs) was 0.2% (2 of 914) in ofatumumab-treated patients and no patients with treatment enhancing or neutralising ADA were identified. The impact of positive ADA titers on PK, safety profile or B-cell kinetics cannot be assessed given the low incidence of ADA associated with ofatumumab.
Clinical efficacy and safety
The efficacy and safety of ofatumumab were evaluated in two randomised, double-blind, active-controlled phase III pivotal studies of identical design (Study 1 [ASCLEPIOS I] and Study 2 [ASCLEPIOS II]) in patients with relapsing forms of MS (RMS) aged 18 to 55 years, a disability status at screening with an Expanded Disability Status Scale (EDSS) score from 0 to 5.5, and who had experienced at least one documented relapse during the previous year or two relapses during the previous two years or positive gadolinium (Gd)-enhancing MRI scan during the previous year. Both newly diagnosed patients and patients switching from their current treatment were enrolled.
In the two studies, 927 and 955 patients with RMS, respectively, were randomised 1:1 to receive either ofatumumab 20 mg subcutaneous injections every 4 weeks starting at week 4 after an initial dosing regimen of three weekly 20 mg doses in the first 14 days (on days 1, 7 and 14) or teriflunomide 14 mg capsules orally once daily. Patients also received matching placebo corresponding to the other treatment arm to ensure blinding (double-dummy design).
The treatment duration for individual patients was variable based on when the end of study criteria were met. Across both studies, the median treatment duration was 85 weeks, 33.0% of patients in the ofatumumab group vs 23.2% of patients in the teriflunomide group were treated more than 96 weeks.
Demographics and baseline characteristics were well-balanced across treatment arms and both studies (see Table 2). Mean age was 38 years, mean disease duration was 8.2 years since onset of first symptom, and mean EDSS score was 2.9; 40% of patients had not been previously treated with a disease-modifying therapy (DMT) and 40% had gadolinium (Gd)-enhancing T1 lesions on their baseline MRI scan.
The primary efficacy endpoint of both studies was the annualised rate of confirmed relapses (ARR) based on EDSS. Key secondary efficacy endpoints included the time to disability worsening on EDSS (confirmed at 3 months and 6 months), defined as an increase in EDSS of ≥1.5, ≥1, or ≥0.5 in patients with a baseline EDSS of 0, 1 to 5, or ≥5.5, respectively. Further key secondary endpoints included the number of Gd-enhancing T1 lesions per MRI scan, the annualised rate of new or enlarging T2 lesions and the neurofilament light chain (NfL) concentration in serum. Disability-related key secondary endpoints were evaluated in a meta-analysis of combined data from ASCLEPIOS Study 1 and Study 2, as defined in the study protocols.
Table 2. Demographics and baseline characteristics:
| Characteristics | Study 1 (ASCLEPIOS I) | Study 2 (ASCLEPIOS II) | ||
|---|---|---|---|---|
| Ofatumumab (N=465) | Teriflunomide (N=462) | Ofatumumab (N=481) | Teriflunomide (N=474) | |
| Age (mean ± standard deviation; years) | 39±9 | 38±9 | 38±9 | 38±9 |
| Sex (female; %) | 68.4 | 68.6 | 66.3 | 67.3 |
| Duration of MS since diagnosis (mean/median; years) | 5.77 / 3.94 | 5.64 / 3.49 | 5.59 / 3.15 | 5.48 / 3.10 |
| Previously treated with DMTs (%) | 58.9 | 60.6 | 59.5 | 61.8 |
| Number of relapses in last 12 months | 1.2 | 1.3 | 1.3 | 1.3 |
| EDSS score (mean/median) | 2.97 / 3.00 | 2.94 / 3.00 | 2.90 / 3.00 | 2.86 / 2.50 |
| Mean total T2 lesion volume (cm³) | 13.2 | 13.1 | 14.3 | 12.0 |
| Patients with Gd+ T1 lesions (%) | 37.4 | 36.6 | 43.9 | 38.6 |
| Number of Gd+ T1 lesions (mean) | 1.7 | 1.2 | 1.6 | 1.5 |
The efficacy results for both studies are summarised in Table 3, Figure 1 and Figure 2.
In both phase III studies, ofatumumab compared to teriflunomide demonstrated a significant reduction in the annualised relapse rate of 50.5% and 58.4%, respectively.
The pre-specified meta-analysis of combined data showed that ofatumumab compared to teriflunomide significantly reduced the risk of 3-month confirmed disability progression (CDP) by 34.3% and the risk of 6-month CDP by 32.4% (see Figure 1).
Ofatumumab compared to teriflunomide significantly reduced the number of Gd-enhancing T1 lesions by 95.9% and the rate of new or enlarging T2 lesions by 83.5% (values represent mean reductions for the combined studies).
Ofatumumab compared to teriflunomide significantly reduced NfL concentrations from the first assessment at 3 months (see Table 3 and Figure 2).
A similar effect of ofatumumab on the key efficacy results compared to teriflunomide was observed across the two phase III studies in exploratory subgroups defined by sex, age, body weight, prior non-steroid MS therapy, and baseline disability and disease activity.
Table 3. Overview of key results from phase III studies in RMS:
| Characteristics | Study 1 (ASCLEPIOS I) | Study 2 (ASCLEPIOS II) | ||
|---|---|---|---|---|
| Ofatumumab 20 mg (N=465) | Teriflunomide 14 mg (N=462) | Ofatumumab 20 mg (N=481) | Teriflunomide 14 mg (N=474) | |
| Endpoints based on separate studies | ||||
| Annualised relapse rate (ARR) (primary endpoint)1 | 0.11 | 0.22 | 0.10 | 0.25 |
| Rate reduction | 50.5% (p<0.001) | 58.4% (p<0.001) | ||
| Mean number of T1 Gd-enhancing lesions per MRI scan | 0.0115 | 0.4555 | 0.0317 | 0.5172 |
| Relative reduction | 97.5% (p<0.001) | 93.9% (p<0.001) | ||
| Number of new or enlarging T2 lesions per year | 0.72 | 4.00 | 0.64 | 4.16 |
| Relative reduction | 81.9% (p<0.001) | 84.6% (p<0.001) | ||
| NfL at 3 months (pg/ml) | 8.80 | 9.41 | 8.92 | 10.02 |
| Relative reduction | 7% (p=0.011) | 11% (p<0.001) | ||
| Endpoints based on pre-specified meta-analyses | ||||
| Proportion of patients with 3-month confirmed disability progression2 Risk reduction | 10.9% ofatumumab vs. 15.0% teriflunomide 34.3% (p=0.003) | |||
| Proportion of patients with 6-month confirmed disability progression2 Risk reduction | 8.1% ofatumumab vs. 12.0% teriflunomide 32.4% (p=0.012) | |||
1 Confirmed relapses (accompanied by a clinically relevant change in the EDSS).
2 Kaplan-Meier estimates at 24 months. 3- and 6-month CDP were assessed based on prospectively planned analysis of the combined data from the two phase III studies and defined as a clinically meaningful increase in the EDSS sustained for at least 3 or 6 months, respectively. A clinically meaningful increase in EDSS is defined as an increase of at least 1.5 points if the baseline EDSS score was 0, an increase of at least 1.0 point if the baseline EDSS score was 1.0–5.0, and an increase of at least 0.5 points if the baseline EDSS score was 5.5 or greater.
Figure 1. Time to first 3-month CDP by treatment (ASCLEPIOS Study 1 and Study 2 combined, full analysis set):
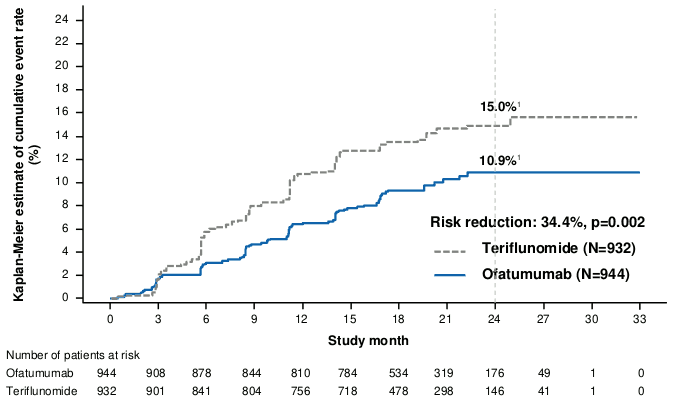
1 The numbers shown on the curves represent Kaplan-Meier estimates of the risk of the event at 24 months (marked by the vertical dashed line).
Figure 2. NfL concentrations in serum by treatment (ASCLEPIOS Study 1 and Study 2 combined, full analysis set):
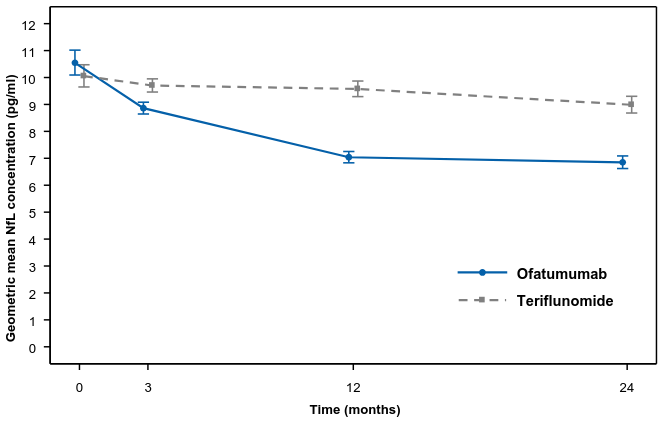
The line plots represent the adjusted geometric means with 95% CI at each time point which are from Repeated measures model. Geometric means at baseline are derived as exponentiated arithmetic mean of natural logarithmic of raw values of NfL concentrations in serum.
In the phase III studies, the proportion of patients with adverse events (AEs) (83.6% vs 84.2%) and the AEs leading to discontinuation (5.7% vs 5.2%) were similar in the ofatumumab and teriflunomide groups.
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with Kesimpta in one or more subsets of the paediatric population in the treatment of multiple sclerosis (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Absorption
After subcutaneous administration, ofatumumab has a prolonged release/absorption profile (Tmax of 4.3 days) and is predominantly absorbed via the lymphatic system.
A monthly subcutaneous dose of 20 mg leads to a mean AUCtau of 483 μg*h/ml and a mean Cmax of 1.43 μg/ml at steady state.
Distribution
The volume of distribution at steady state was estimated to be 5.42 litres following repeated subcutaneous administration of ofatumumab at a dose of 20 mg.
Biotransformation
Ofatumumab is a protein for which the expected metabolic pathway is degradation to small peptides and amino acids by ubiquitous proteolytic enzymes.
Elimination
Ofatumumab is eliminated in two ways: a target-mediated route that is related to binding to B cells and a target-independent route mediated by non-specific endocytosis followed by intracellular catabolism, as with other IgG molecules. B cells present at baseline result in a greater component of target-mediated clearance of ofatumumab at the start of therapy. Ofatumumab dosing leads to potent depletion of B cells resulting in reduced overall clearance.
The half-life at steady state was estimated to be approximately 16 days following repeated subcutaneous administration of ofatumumab at a dose of 20 mg.
Linearity/non-linearity
Ofatumumab had non-linear pharmacokinetics related to its decreasing clearance over time.
Special populations
Adults over 55 years old
There are no dedicated pharmacokinetic studies of ofatumumab in patients over 55 years old due to limited clinical experience (see section 4.2).
Paediatric population
No studies have been conducted to investigate the pharmacokinetics of ofatumumab in paediatric patients below the age of 18 years.
Gender
Gender had a modest (12%) effect on ofatumumab central volume of distribution in a cross-study population analysis, with higher Cmax and AUC values observed in female patients (48% of the patients in this analysis were male and 52% were female); these effects are not considered clinically relevant, and no dose adjustment is recommended.
Body weight
Based on the results of a cross-study population analysis, body weight was identified as a covariate of exposure (Cmax and AUC) to ofatumumab in RMS subjects. However, body weight did not affect safety and efficacy measures evaluated in the clinical studies; therefore, dose adjustment is not required.
Renal impairment
No specific studies of ofatumumab in patients with renal impairment have been performed.
Patients with mild renal impairment were included in clinical studies. There is no experience in patients with moderate and severe renal impairment. However, as ofatumumab is not excreted via urine, it is not expected that patients with renal impairment require dose modification.
Hepatic impairment
No studies of ofatumumab in patients with hepatic impairment have been performed.
Since hepatic metabolism of monoclonal antibodies such as ofatumumab is negligible, hepatic impairment is not expected to impact its pharmacokinetics. Therefore, it is not expected that patients with hepatic impairment require dose modification.
5.3. Preclinical safety data
Non-clinical data revealed no special hazard for humans based on conventional studies of repeated dose toxicity including safety pharmacology endpoints.
Neither carcinogenicity nor mutagenicity studies have been conducted with ofatumumab. As an antibody, ofatumumab is not expected to interact directly with DNA.
The embryo-foetal development (EFD) and the enhanced pre/post-natal development (ePPND) studies in monkeys showed that exposure to ofatumumab given intravenously during gestation caused no maternal toxicity, no teratogenicity, and no adverse effects on embryo-foetal and pre/post-natal development.
In these studies, ofatumumab was detected in the blood of the foetuses and infants, confirming placental transfer and foetal exposure to ofatumumab persisting post-natally (long half-life of the monoclonal antibody). Exposure to ofatumumab during gestation led to the expected depletion of CD20+ B cells in maternal animals and their foetuses and infants, along with a reduced spleen weight (without histological correlate) in foetuses and a reduced humoral immune response to keyhole limpet haemocyanin (KLH) in infants at high doses. All these changes were reversible during the 6-month post-natal period. In infants, early post-natal mortality was observed at a dose 160 times higher than the therapeutic dose (on AUC basis) and was likely due to potential infections secondary to immunomodulation. The NOAEL related to the pharmacological activity of ofatumumab in infants of the ePPND study leads to an AUC-based safety margin of at least 22-fold when maternal exposure at the NOAEL is compared with human exposure at the therapeutic dose of 20 mg monthly.
In a dedicated monkey fertility study, male and female fertility endpoints were unaffected.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.