KIENDRA Film-coated tablet Ref.[115230] Active ingredients: Siponimod
Source: Health Products Regulatory Authority (ZA) Revision Year: 2023 Publisher: Novartis SA (Pty) Ltd., Magwa Crescent West, Waterfall City, Johannesburg, 2090
Pharmacodynamic properties
Pharmacotherapeutic group: Selective immunosuppressants
ATC code: L04AA42
Siponimod is a sphingosine-1-phosphate (S1P) receptor modulator. Siponimod binds selectively on two out of five G-protein-coupled receptors (GPCRs) for S1P, namely S1P1 and S1P5. By acting as a functional antagonist on S1P1 receptors on lymphocytes, siponimod prevents egress from lymph nodes. This reduces the recirculation of T-cells into the central nervous system (CNS) to limit central inflammation. Siponimod spares effector memory T-cells in peripheral tissues and blood and does not impair lymphocyte activation.
Siponimod readily crosses the blood brain barrier.
In animal studies direct effects have been demonstrated for siponimod on neural cells, via S1P1 on astrocytes and S1P5 on oligodendrocytes. In a mouse model of experimental autoimmune encephalomyelitis a direct neuroprotective effect, independent from effects on lymphocytes, was also demonstrated for siponimod applied centrally (via intracerebroventricular infusions).
Immune system
Siponimod induces a dose-dependent reduction of the peripheral blood lymphocyte count within 6 hours of the first dose, due to the reversible sequestration of lymphocytes in lymphoid tissues.
With continued daily dosing, the lymphocyte count continues to decrease, reaching a nadir median (90% CI) lymphocyte count of approximately 0.560 (0.271 to 1.08) cells/nL in a typical CYP2C9*1*1 or *1*2, non-Japanese SPMS patient, corresponding to 20 to 30% of baseline. Low lymphocyte counts are maintained with chronic daily dosing.
Before initiating treatment, a recent complete blood count (CBC) (i.e. within last 6 months or after discontinuation of prior therapy) should be available. Absolute lymphocyte counts <0.2 x 109/l, if confirmed, should lead to dose reduction to 1 mg, because in clinical studies siponimod dose was reduced in patients with absolute lymphocyte counts <0.2 x 109/l. Confirmed absolute lymphocyte counts <0.2 x 109/l in a patient already receiving siponimod 1 mg should lead to interruption of siponimod therapy until the level reaches 0.6 x 109/l when re-initiation of siponimod can be considered.
Lymphocyte counts typically return to the normal range in the vast majority (90%) of SPMS patients within 10 days of stopping therapy. After stopping siponimod treatment residual lowering effects on peripheral lymphocyte count may persist for up to 3 to 4 weeks after the last dose.
Cardiac electrophysiology
Heart rate and rhythm
Siponimod causes a transient reduction in heart rate and atrioventricular conduction upon treatment initiation (see section 4.8). The maximum decline in heart rate is seen in the first 6 hours post-dose. Autonomic responses of the heart, including diurnal variation of heart rate and response to physical exercise, are not affected by siponimod treatment.
A transient, dose-dependent decrease in heart rate was observed during the initial dosing phase of siponimod, that plateaued at doses ≥5 mg and bradyarrhythmic events (AV Blocks and sinus pauses) were detected at a higher incidence under siponimod treatment compared to placebo.
No second degree AV blocks of Mobitz type II or higher degree were observed. Most AV blocks and sinus pauses occurred above the therapeutic dose of 2 mg with notably higher incidence under non titrated conditions compared to dose titration conditions.
The decrease in heart rate induced by siponimod can be reversed by atropine or isoprenaline.
Potential to prolong the QT interval
The effects of therapeutic (2 mg) and supratherapeutic (10 mg) doses of siponimod on cardiac repolarization were investigated in a thorough QT study. The results did not suggest an arrhythmogenic potential related to QT prolongation with siponimod. Siponimod increased the mean placebo-corrected baseline-adjusted mean QTcF (ΔΔQTcF) by more than 5 ms with a maximum mean effect of 7.8 ms (2 mg) and 7.2 ms (10 mg), respectively at 3 h post-dose. The upper bound of the one-sided 95% CI for the ΔΔQTcF at all time points remained below 10 ms. Categorical analysis revealed no treatment-emergent QTc values above 480 ms, no QTc increases from baseline of more than 60 ms and no corrected or uncorrected QT/QTc value exceeded 500 ms.
Pulmonary function
Siponimod treatment with single doses or multiple doses for 28 days is not associated with clinically relevant increases in airway resistance as measured by forced expiratory volume in 1 second (FEV1) and forced expiratory flow (FEF) during expiration of 25 to 75% of the forced vital capacity (FEF25-75).
A slight trend of reduced FEV1 was detected at non-therapeutic single doses (>10 mg). Multiple doses of siponimod were associated with mild to moderate changes in FEV1 and FEF25-75% which were not dose and daytime dependent and were not associated with any clinical signs of increased airway resistance.
Concomitant treatment of siponimod with propranolol resulted in minimal decrease of FEV1 in comparison to propranolol alone. The changes with the individual medicines or with the combination were within the physiological variability of FEV1 and not clinically significant.
Clinical studies
Study A2304 (EXPAND) in SPMS
Study A2304 was a randomised, double-blind, placebo-controlled, event and follow-up duration driven, phase 3 study in patients with SPMS who had documented evidence of progression in the prior 2 years in the absence or independent of relapses, no evidence of relapse in 3 months prior to study enrollment and with Expanded Disability Status Scale (EDSS) score of 3.0 to 6.5 at study entry.
Patients were randomised 2:1 to receive either once daily siponimod 2 mg or placebo. Evaluations were performed at screening and every 3 months and at the time of relapse. MRI evaluations were performed at screening and every 12 months.
The primary endpoint of the study was the time to 3-month confirmed disability progression (CDP) determined as at least a 1-point increase from baseline in EDSS (0.5 point increase for patients with baseline EDSS of 5.5 or more) sustained for 3 months. Key secondary endpoints were time to 3-month confirmed worsening of at least 20% from baseline in the timed 25-foot walk test (T25FW) and change from baseline in T2 lesion volume. Additional secondary endpoints included time to 6-month CDP, percent brain volume change, measures of inflammatory disease activity.
Study duration was variable for individual patients (median study duration was 21 months, range 1 day to 37 months).
Median age was 49.0 years, median disease duration was 16.0 years and median EDSS score was 6.0 at baseline; 63.9% of patients had no relapses in the 2 years prior to study entry and 78% had no gadolinium (Gd)-enhancing lesions on their baseline MRI scan; 78.3% of patients had been previously treated with a therapy for their MS.
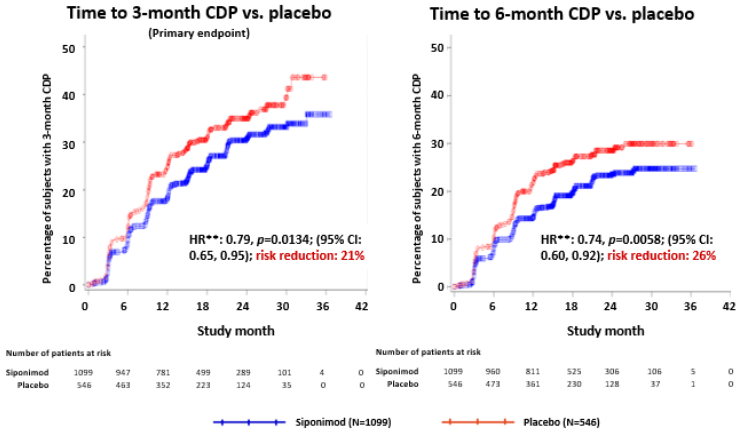
Time to onset of 3-month confirmed disability progression (primary endpoint) was significantly delayed for siponimod with a 21.2% risk reduction compared to placebo (hazard ratio (HR) 0.79, p<0.0134).
The results for this study are summarized in Table 3 and Figure-1.
Table 3. Overview of results from efficacy endpoints:
| Efficacy Parameter | Statistic | Estimate (95% CI) | p-value |
|---|---|---|---|
| Clinical | |||
| Time to 3-month CDP (primary endpoint) | Hazard ratio1 | 0.79 (0.65,0.95) | 0.0134 |
| Time to 6-month CDP | Hazard ratio1 | 0.74 (0.60, 0.92) | 0.0058 |
| Annualized relapse rate (ARR), confirmed relapses | ARR ratio3 | 0.45 (0.34, 0.59) | <0.0001 |
| Change from baseline in Symbol Digit Modality Test | Treatment difference4 | 1.38 (0.58, 2.18) | 0.0007 |
| MRI | |||
| Change from baseline in T2 lesion volume (mm³) | Treatment difference2 | -695 (-877, -513) | <0.0001 |
| Percent brain volume change relative to baseline | Treatment difference2 | 0.15 (0.07,0.23) | 0.0002 |
| Number of Gd-enhancing T1 weighted lesions | Rate ratio5 | 0.14 (0.10, 0.19) | <0.0001 |
| Number of new/enlarging T2 lesions | Rate ratio6 | 0.19 (0.16, 0.24) | <0.0001 |
All analyses are based on the full analysis set (FAS), which includes all randomized subjects who took at least one dose of study medication. p values are two-sided.
1 Hazard ratio (siponimod/placebo), Cox proportional hazard model.
2 Treatment difference in the average over mean changes at Months 12 and 24, repeated measures model.
3 ARR ratio (siponimod/placebo), negative binomial model.
4 Treatment difference in the average over all visits, repeated measures model.
5 Rate ratio (siponimod/placebo) up to and including Month 24, negative binomial model.
6 Rate ratio (siponimod/placebo) per scan, average over Months 12 and 24, negative binomial model.
Figure 1. Patients with 3-month and 6-month CDP based on EDSS-Kaplan-Meier curves (FAS):
Siponimod did not significantly delay time to 3-month confirmed ≥20% deterioration in the T25FW (a numerical 6.2% risk reduction was observed).
Results from the study showed a consistent risk reduction in the time to 3-month CDP with siponimod in subgroups defined based on gender, age, prior multiple sclerosis therapy, pre-study relapse activity, baseline MRI disease activity and disability levels at baseline.
Pharmacokinetic properties
Absorption
The time (Tmax) to reach maximum plasma concentrations (Cmax) after multiple oral administration of siponimod was about 4 hours (range 2 to12 hours). Siponimod absorption is extensive (≥70%, based on the amount of radioactivity excreted in urine and the amount of metabolites in feces extrapolated to infinity). The absolute oral bioavailability of siponimod is approximately 84%. For 2 mg siponimod given once daily over 10 days, a mean Cmax of 30.4 ng/mL and mean AUCtau of 558 h*ng/mL were observed on day 10. Steady state was reached after approximately 6 days of multiple once daily administration of siponimod.
Food effect
Food intake had no effect on the systemic exposure of siponimod (Cmax and AUC). Therefore KIENDRA may be taken without regard to meals (see section 4 Dosage regimen and administration).
Distribution
Siponimod is distributed to body tissues with a moderate mean volume of distribution of 124 L. Siponimod fraction found in plasma is 68% in humans. Animal studies show that siponimod readily crosses the blood-brain-barrier. Protein binding of siponimod is >99.9% in healthy subjects and in hepatic and renal impaired patients.
Biotransformation/metabolism
Siponimod is extensively metabolised, mainly via CYP2C9 (79.3%), followed by CYP3A4 (18.5%). The pharmacological activity of the main metabolites M3 and M17 is not expected to contribute to the clinical effect and the safety of siponimod in humans.
Elimination
An apparent systemic clearance (CL/F) of 3.11 L/h was estimated in MS patients. The apparent elimination half-life is approximately 30 hours. Siponimod is eliminated from the systemic circulation mainly due to metabolism, and subsequent biliary/fecal excretion. Unchanged siponimod was not detected in urine.
Linearity/non-linearity
Siponimod concentration increases in an apparent dose proportional manner after multiple once daily doses of siponimod 0.3 mg to 20 mg.
Steady-state-plasma concentrations are reached after approximately 6 days of once daily dosing and steady-state levels are approximately 2 to 3-fold greater than the initial dose. An up-titration regimen is used to stepwise reach the clinical therapeutic dose of siponimod of 2 mg after 6 days and 4 additional days of dosing are required to reach the steady-state-plasma concentrations.
In vitro and in vivo evaluation of medicine interaction potential
Siponimod (and metabolites M3, M17) as a causative medicine of interaction. In vitro investigations indicated that siponimod and its major systemic metabolites M3 and M17 do not show any clinically relevant medicine-medicine interaction potential at the therapeutic dose of 2 mg once daily for all investigated CYP enzymes and transporters, and do not necessitate clinical investigation.
Siponimod as a medicine of interaction
CYP2C9 is polymorphic and the genotype influences the fractional contributions of the two oxidative metabolism pathways to overall elimination. Physiologically based pharmacokinetic modeling indicates a differential CYP2C9 genotype-dependent inhibition and induction of CYP3A4 pathways. With decreased CYP2C9 metabolic activity in the respective genotypes, a larger effect of the CYP3A4 perpetrators on siponimod exposure is anticipated.
Co-administration of siponimod with CYP2C9 and CYP3A4 inhibitors
The co-administration of fluconazole (moderate CYP2C9/CYP3A4 inhibitor) 200 mg daily at steady-state and a single dose of siponimod 4 mg in CYP2C9*1*1 healthy volunteers led to a two-fold increase in the AUC of siponimod. Mean siponimod terminal half-life was increased by 50%.
Co-administration of siponimod with CYP2C9 and CYP3A4 inducers
Strong CYP3A4/moderate 2C9 inducers (e.g. carbamazepine) and moderate CYP3A4 inducers (e.g. modafinil) significantly reduced siponimod AUC by up to 76% and up to 51%, respectively, according to clinical interaction studies and in silico evaluation of the medicine interaction potential. The co- administration of siponimod 2 mg daily in the presence of 600 mg daily doses of rifampin (strong CYP3A4 and moderate CYP2C9 inducer) decreased siponimod AUCtau,ss and Cmax,ss by 57% and 45%, respectively in CY2C9*1*1 subjects.
Special populations
Elderly patients (65 years or above)
Results from population pharmacokinetics suggest that dose adjustment would not be necessary in elderly patients. However, to date clinical experience in patients aged above 65 years is limited.
Gender
Gender has no influence on siponimod pharmacokinetics.
Renal impairment
No siponimod dose adjustments are needed in patients with mild, moderate or severe renal impairment. Mean siponimod half-life and Cmax (total and unbound) were comparable between subject with severe renal impairment and healthy subjects. Total and unbound AUCs were only slightly increased (by 23 to 33%), compared to healthy subjects. The effects of end-stage renal disease or hemodialysis on the pharmacokinetics of siponimod has not been studied. Due to the high plasma protein binding (>99.9%) of siponimod, haemodialysis is not expected to alter the total and unbound siponimod concentration and no dose adjustments are anticipated based on these considerations.
Hepatic impairment
No dose adjustments for siponimod are needed in patients with hepatic impairment. The unbound siponimod pharmacokinetics AUC is 15% and 50% higher in subjects with moderate and severe hepatic impairment, respectively, in comparison with healthy subjects for the 0.25 mg single dose studied. The mean half-life of siponimod was unchanged in hepatic impairment.
Pharmacogenomics
The CYP2C9 genotype has a significant impact on siponimod metabolism. Patients homozygous for CYP2C9*3 (CYP2C9*3*3 genotype: approximately 0.3 to 0.4% of Caucasians and less in others) are contraindicated with siponimod (see section 4.3). Use of siponimod in these patients results in substantially elevated siponimod plasma levels. The recommended maintenance dose of siponimod is 1 mg daily in patients with CYP2C9 *2*3 or *1*3 genotype to avoid an increased exposure to siponimod (see sections 4.2).
There are other less frequent occurring polymorphisms for CYP2C9. The pharmacokinetics of siponimod have not been evaluated in such subjects. Some polymorphisms such as *5, *6, *8 and *11 are associated with decreased or loss of enzyme function. It is estimated that CYP2C9 *5, *6, *8 and *11 alleles have a combined frequency of approximately 10% in populations with African ancestry, 2% in Latinos/Hispanics and <0.4% in Caucasians and Asians.
After a single dose of 0.25 mg siponimod, AUCinf and AUClast was approximately 2- and 4-fold higher in subjects with the CYP2C9*2*3 and CYP2C9*3*3 genotypes, respectively, while there was only a minor increase of Cmax by 21% and 16%, respectively, compared to extensive metabolizers (CYP2C9*1*1). The mean half-life was prolonged in CYP2C9*2*3 and CYP2C9*3*3 carriers (51 and 126 h).
An apparent systemic clearance (CL/F) of about 3.11 L/h was estimated in CYP2C9 extensive metabolizer (CYP2C9*1*1 and CYP2C9*1*2) SPMS patients after multiple oral administrations of siponimod. Cl/F is 2.5, 1.9, 1.6, and 0.9 L/h in subjects with the CYP2C9*2*2, CYP2C9*1*3, CYP2C9*2*3 and CYP2C9*3*3 genotypes, respectively. The resultant increase in siponimod AUC was 25, 61, 91, 285% in subjects with the CYP2C9*2*2, CYP2C9*1*3, CYP2C9*2*3 and CYP2C9*3*3 genotypes, respectively, as compared to those with the CYP2C9*1*1 genotype. As the apparent clearance estimated for subjects with the CYP2C9*1*2 genotype was comparable to that for subjects of the CYP2C9*1*1 genotype, similar siponimod exposure is expected for both genotypes.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.