KYNTHEUM Solution for injection Ref.[6630] Active ingredients: Brodalumab
Source: European Medicines Agency (EU) Revision Year: 2018 Publisher: LEO Pharma A/S, Industriparken 55, DK-2750, Ballerup, Denmark
Pharmacodynamic properties
Pharmacotherapeutic group: Immunosuppressants, Interleukin inhibitors
ATC code: L04AC12
Mechanism of action
Brodalumab is a recombinant fully human monoclonal immunoglobulin IgG2 antibody that binds with high affinity to human IL-17RA and blocks the biological activities of the pro-inflammatory cytokines IL-17A, IL-17F, IL-17A/F heterodimer and IL-25, resulting in inhibition of the inflammation and clinical symptoms associated with psoriasis. IL-17RA is a protein expressed on the cell surface and is a required component of receptor complexes utilized by multiple IL-17 family cytokines. IL-17 family cytokine concentrations have been reported to be increased in psoriasis. IL-17A, IL-17F and IL-17A/F heterodimer have pleiotropic activities including the induction of pro-inflammatory mediators such as IL-6, GROα, and G-CSF from epithelial cells, endothelial cells and fibroblasts that promote tissue inflammation. Blocking IL-17RA inhibits IL-17 cytokine-induced responses resulting in normalization of inflammation in the skin.
Pharmacodynamic effects
Elevated levels of IL-17A, IL-17C and IL-17F gene expression are found in psoriatic plaques. Elevated levels of expression of IL-12B and IL-23A, the genes for the two subunits of IL-23, an upstream activator of IL-17A and IL-17F expression, are also found in psoriatic plaques. Treatment with Kyntheum in psoriasis patients has been shown to decrease levels of IL-17A and markers of cell proliferation and epidermal thickness in lesional skin biopsies to non-lesional skin biopsy levels up to 12 weeks post-treatment.
Clinical efficacy and safety
The efficacy and safety of Kyntheum was assessed in 4373 adult plaque psoriasis patients across three multinational, randomised, double-blind, phase 3, placebo-controlled clinical trials (AMAGINE-1, AMAGINE-2, and AMAGINE-3). AMAGINE-2 and AMAGINE-3 were also active comparator (ustekinumab)-controlled. All three trials included a 12-week placebo-controlled induction phase, a double-blind duration of 52 weeks, and an open-label long-term extension.
Patients enrolled were candidates for systemic therapy, including phototherapy, biologic and nonbiologic systemic therapies. Approximately 21% of patients had a history of psoriatic arthritis. Approximately 30% of patients had previously received a biological and 12% of patients were biological failures.
Patients were predominantly men (69%) and white (91%), with a mean age of 45 years (18 to 86 years), of these 6.1% were >65 years of age and 0.3% were >75 years of age. Across treatment groups, the baseline Psoriasis Area Severity Index (PASI) score ranged from 9.4 to 72 (median: 17.4) and baseline involved body surface area (BSA) ranged from 10 to 97 (median: 21). Baseline static Physician Global Assessment (sPGA) score ranged from "3 (moderate)" (58%) to "5 (very severe)" (5%).
AMAGINE-1 was conducted in 661 patients. The trial included a 12-week double-blind, placebocontrolled induction phase followed by a double-blind withdrawal and retreatment phase up to 52 weeks. Patients randomised to Kyntheum received 210 mg or 140 mg at Week 0 (day 1), Week 1, and Week 2 followed by same dose every 2 weeks. At Week 12, patients originally randomised to Kyntheum who achieved sPGA success (0 or 1) were re-randomised to receive either placebo or continued Kyntheum at their induction dose. Patients originally randomised to placebo and those who did not meet the criteria for re-randomisation received Kyntheum 210 mg every two weeks beginning at Week 12. Retreatment was available at or after Week 16 for patients with return of disease and rescue treatment was available after 12 weeks of retreatment.
AMAGINE-2 and AMAGINE-3 were identical placebo- and ustekinumab-controlled trials conducted in 1831 and 1881 patients, respectively. Both trials included a 12-week double-blind, placebo- and ustekinumab-controlled induction phase followed by a double-blind maintenance phase up to 52 weeks. Patients randomised to Kyntheum in the induction phase received 210 mg or 140 mg at Week 0 (day 1), Week 1, and Week 2 followed by same dose every 2 weeks. Patients randomised to ustekinumab received 45 mg for patients ≤100 kg and 90 mg for patients >100 kg at Weeks 0, 4, and 16 followed by same dose every 12 weeks. At Week 12, patients originally randomised to Kyntheum were re-randomised to receive either 210 mg every 2 weeks, or 140 mg every 2 weeks, or 140 mg every 4 weeks, or 140 mg every 8 weeks during the maintenance phase. Patients originally randomised to placebo received Kyntheum 210 mg every 2 weeks beginning at Week 12. At Week 12, patients in the ustekinumab group continued to receive ustekinumab and then were switched to Kyntheum 210 mg every 2 weeks at Week 52. Rescue treatment was available at or after Week 16 for patients with an inadequate response single sPGA ≥3 or persistent sPGA of 2 over at least a 4-week period.
Table 2. Overview of the main efficacy results:
| AMAGINE-1 | AMAGINE-2 and AMAGINE-3 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Placebo | Kyntheum 210 mg Q2W | Placebo | Kyntheum 210 mg Q2W | Ustekinumab | n-randomised | 220 | 222 | 624 | 1236 | 613 | |
| n-completed Week 12 | 209 | 212 | 601 | 1205 | 594 | ||||||
| n-in maintenance | 84 | 83 | NA | 339 | 590 | ||||||
| n-completed Week 52 | 2 | 74 | NA | 236 | 300 | ||||||
| PASI | |||||||||||
| PASI Baseline score (mean±SD) | 19.7±7.7 | 19.4±6.6 | 20.2±8.4 | 20.3±8.3 | 20.0±8.4 | ||||||
| PASI 75 Week 12 (%) | 3 | 83* | 7 | 86* | 70* | ||||||
| PASI 75 Week 52 (%) | 0 | 87* | NA | 65 | 48 | ||||||
| sPGA (%) | |||||||||||
| sPGA 0 or 1 Week 12 | 1 | 76* | 4 | 79* | 59* | ||||||
| sPGA 0 or 1 Week 52 | 0 | 83* | NA | 65 | 45 | ||||||
| PSI | |||||||||||
| PSI Baseline score (mean±SD) | 19.0±6.7 | 18.9±6.7 | 18.8±6.9 | 18.7±7.0 | 18.8±6.9 | ||||||
| PSI responder Week12 (%) | 4 | 61* | 7 | 64* | 54* | ||||||
Q2W = every 2 weeks
PSI = Psoriasis Symptom Inventory. PSI responder: total score ≤8 with no item scores >1; SD: standard
deviation.
Non-responder imputation is used to impute missing data.
Due to re-randomisation to other explored dose regimens, n-in maintenance is substantially lower than n-randomised in several arms. The maintenance phase in AMAGINE-2 and -3 did not include placebo.
* p-value vs. corresponding placebo, adjusted for stratification factors <0.001
PASI 75 response at 2 weeks ranged between 20% and 25% in the Phase 3 trials compared to placebo (0% to 0.6%) and ustekinumab (3% to 3.5%).
Figure 1. PASI 100 during induction and maintenance phase for Kyntheum and ustekinumab (AMAGINE-2 and AMAGINE-3, pooled):
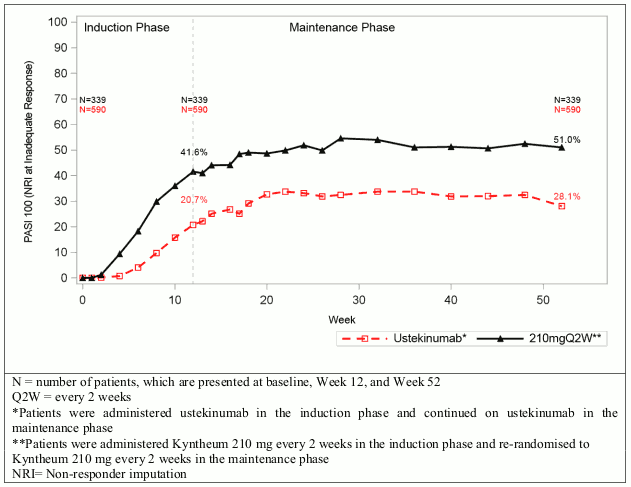
In all three clinical trials, examination of age, gender, race, use of prior systemic or photo therapy, use of prior biologics, and biologic failures did not identify differences in response in all key endpoints [PASI 75, PASI 100, sPGA success (0 or 1), and sPGA clear (0)] to Kyntheum among these subgroups.
Along with primary efficacy endpoints, clinically important improvements were observed in Psoriasis Scalp Severity Index (PSSI) at Week 12 (AMAGINE-1) and in Nail Psoriasis Severity Index (NAPSI) at Week 12 and 52 (AMAGINE-1,-2, and -3).
Quality of life/patient reported outcomes
The proportion of patients who achieved a Psoriasis Symptom Inventory (PSI) score of 0 (not at all) or 1 (mild) on every item (itch, burning, stinging, pain, redness, scaling, cracking and flaking) at Week 12 are shown in Table 2.
The percentage of patients that at Week 12 achieved a DLQI (Dermatology Life Quality Index) score of 0 or 1 was 56%, 61%, 59% in the Kyntheum 210 mg group and 5%, 5%, 7% in the placebo group in AMAGINE-1, -2 and -3, respectively (adjusted p-value<0.001) and 44% in the ustekinumab groups (AMAGINE-2 and -3).
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of trials with Kyntheum in plaque psoriasis in one or more subsets of the paediatric population (see section 4.2 for information on paediatric use).
Pharmacokinetic properties
Absorption
Based on population pharmacokinetic modelling, the estimated accumulation ratio after 20 weeks of dosing is 2.5-fold. In moderate to severe plaque psoriasis patients following a single subcutaneous administration of Kyntheum at 210 mg, the mean maximum serum concentration (Cmax) was 13.4 mcg/ml (standard deviation [SD] = 7.29 mcg/ml). The median time to maximum concentration (Tmax) was 3.0 days (range: 2.0 to 4.0 days) and the mean area under the concentration time curve to the last measurable concentration (AUClast) was 111 mcg*day/ml (SD = 64.4 mcg*day/ml). The subcutaneous bioavailability of brodalumab estimated by population pharmacokinetic modelling was 54.7% (relative standard error [RSE] = 4.25%).
The observed pharmacokinetic parameters during steady-state (weeks 10-12) were: mean steady-state area under the concentration time curve over the dosing interval (AUCtau) was 227.4 mcg*day/ml (SD = 191.7 mcg*day/ml) corresponding to average concentration (Cav,ss) of 16.2 mcg/ml, mean Cmax was 20.9 mcg/ml (SD = 17.0 mcg/ml) and Week 12 mean minimum serum concentration (Ctrough) was 9.8 mcg/ml (SD = 11.2 mcg/ml).
Distribution
Based on population pharmacokinetic modelling, the estimated mean steady-state volume of distribution of brodalumab was approximately 7.24 L.
Biotransformation
As an IgG2 human monoclonal antibody brodalumab is expected to be degraded into small peptides and amino acids via catabolic pathways in a manner similar to endogenous IgG.
Elimination
Following subcutaneous administrations of 210 mg, brodalumab exhibits non-linear pharmacokinetics typical for a monoclonal antibody that undergoes target-mediated drug disposition.
Brodalumab clearance decreases with increasing dose and exposure increases in a greater than dose- proportional manner. For a 3-fold increase in SC brodalumab dose from 70 to 210 mg, the steady-state serum brodalumab Cmax and AUC0-t increased approximately 18- and 25-fold, respectively.
Following a single subcutaneous administration of brodalumab 210 mg in plaque psoriasis patients, the apparent clearance (CL/F) is 2.95 L/day.
Population pharmacokinetic modelling predicted that serum brodalumab concentrations dropped below the quantification limit (0.05 mcg/ml) 63 days after discontinuation of steady-state dosing of brodalumab 210 mg administered every 2 weeks in 95% of the patients. However, brodalumab concentrations below LLOQ (Lower Limit of Quantification) were associated with IL-17 receptor occupancy up to 81%.
Based on population pharmacokinetic modelling the estimated half-life of brodalumab was 10.9 days at steady-state after every other week subcutaneous dose of 210 mg.
Impact of weight on pharmacokinetics
Population pharmacokinetic modelling indicated that exposure decreased as body weight increased. No dose adjustment is recommended.
Elderly patients
Population pharmacokinetic modelling indicated that age did not have an effect on brodalumab pharmacokinetics, which was co-based on 259 (6%) patients being 65-74 years old and on 14 (0.3%) patients being ≥75 years old, within a total PK population of 4271 plaque psoriasis patients.
Renal or hepatic impairment
No pharmacokinetic data are available in patients with impaired renal or hepatic function. Renal elimination of intact brodalumab, an IgG monoclonal antibody, is expected to be low and of minor consequence. Brodalumab is expected to be mainly eliminated via catabolism and hepatic impairment is not expected to influence clearance.
Other populations
The pharmacokinetics of brodalumab was similar between Japanese and non-Japanese patients with psoriasis.
Population pharmacokinetic analysis indicated that gender did not have an effect on brodalumab pharmacokinetics.
Pharmacokinetic/pharmacodynamic relationship(s)
A population pharmacokinetic/pharmacodynamic model, developed using all available data indicated that at a dose of 210 mg every 2 weeks, 90% of all patients would be predicted to maintain a trough concentration greater than the estimated IC90 value of 1.51 mcg/ml. Based on an exploratory descriptive analysis, no relationship was observed between exposure and incidence of serious infections and infestations, candida infections, viral infections, and suicidal ideation and behaviour events. Exposure-response analysis indicates that higher brodalumab concentrations are related to better PASI and sPGA response.
Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of repeated dose toxicity (including safety pharmacology endpoints and assessment of fertility-related endpoints), and toxicity to reproduction and development.
Carcinogenicity studies with brodalumab have not been conducted. However, there were no proliferative changes in cynomolgus monkeys administered weekly subcutaneous doses of brodalumab at 90 mg/kg for 6 months (AUC exposure 47-fold higher than in human patients receiving Kyntheum 210 mg every 2 weeks). The mutagenic potential of brodalumab was not evaluated; however, monoclonal antibodies are not expected to alter DNA or chromosomes.
In cynomolgus monkeys there were no effects on male and female reproductive organs and on sperm count, motility and morphology following administration of brodalumab at dose levels up to 90 mg/kg once weekly for 6 months, (AUC exposure up to 47-fold higher than in human patients receiving Kyntheum 210 mg every 2 weeks).
In cynomolgus monkeys, no effects on embryo-foetal or postnatal (up to 6 months of age) development were observed when brodalumab was dosed subcutaneously throughout pregnancy at exposure levels up to 27-fold higher than those achieved in human patients receiving Kyntheum 210 mg every 2 weeks based on the area under the concentration curve (AUC). Serum concentrations in monkey infants and in foetal rabbits indicated considerable passage of brodalumab from the mother to the foetus at the end of pregnancy.
In cynomolgus monkeys, after weekly subcutaneous dosing of brodalumab at dose levels up to 90 mg/kg for 6 months, brodalumab-related effects were limited to injection site reactions and mucocutaneous inflammation that was consistent with pharmacologic modulation of host surveillance to commensal microflora. There were no effects on peripheral blood immunophenotyping and the Tcell dependent antibody response assay. In a local tolerance test in rabbits, moderate to severe edema was observed after subcutaneous injection of a formulation containing brodalumab at the clinical concentration of 140 mg/ml.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.