LARTRUVO Concentrate for solution for infusion Ref.[8998] Active ingredients: Olaratumab
Source: European Medicines Agency (EU) Revision Year: 2019 Publisher: Eli Lilly Nederland B.V., Papendorpseweg 83, 3528 BJ Utrecht, The Netherlands
Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic agents, monoclonal antibodies
ATC code: L01XC27
Mechanism of action
Olaratumab is an antagonist of platelet derived growth factor receptor-α (PDGFR-α), expressed on tumour and stromal cells. Olaratumab is a targeted, recombinant, fully human immunoglobulin G subclass 1 (IgG1) monoclonal antibody that specifically binds PDGFR-α, blocking PDGF AA, -BB, and -CC binding and receptor activation. As a result, in vitro olaratumab inhibits PDGFR-α pathway signalling in tumour and stromal cells. In addition, in vivo olaratumab has been shown to disrupt the PDGFR-α pathway in tumour cells and inhibit tumour growth.
Immunogenicity
As with all therapeutic proteins, there is the potential for immunogenicity.
Overall, a low incidence of both treatment emergent anti-drug antibodies and neutralising antibodies were detected in clinical trial samples.
Clinical efficacy and safety
The efficacy and safety of olaratumab was assessed in a Phase 1b/2, multi-centre study in anthracycline naïve patients with histologically or cytologically confirmed, advanced soft tissue sarcoma not amenable to receive surgery or radiotherapy with curative intent. Patients with gastrointestinal stromal tumours (GIST) or Kaposi sarcoma were not enrolled. The Phase 2 portion of the study was a randomised, open label study of olaratumab plus doxorubicin versus doxorubicin alone. A total of 133 patients were randomised, of whom 129 received at least one dose of study treatment (64 in the olaratumab plus doxorubicin arm and 65 in the doxorubicin arm). Patients were required to have histologically or cytologically confirmed, advanced soft tissue sarcoma and ECOG performance status of 0-2. Randomisation was stratified by PDGFR-α expression (positive versus negative), number of previous lines of treatment (0 versus 1 or more lines), histological tumour type (leiomyosarcoma, synovial sarcoma, and others) and ECOG performance status (0 or 1 versus 2).
Patients were randomised in a 1:1 ratio to either olaratumab (15 mg/kg) on Day 1 and Day 8 plus doxorubicin (75 mg/m²) on Day 1 of each 21-day cycle for up to 8 cycles or doxorubicin (75 mg/m²) alone on Day 1 of each 21-day cycle, also for up to 8 cycles. Olaratumab and doxorubicin were administered by intravenous infusion. During Cycles 5 to 8 on both arms, dexrazoxane (dosed at a ratio of 10:1 to the administered dose of doxorubicin) could be administered on Day 1 of each cycle at the investigator's discretion to reduce potential doxorubicin-related cardiotoxicity. All patients receiving more than 4 cycles of doxorubicin received dexrazoxane. Patients in the olaratumab plus doxorubicin arm could continue on olaratumab monotherapy until disease progression, unacceptable toxicity or any other reason for treatment discontinuation occurred.
Demographics and baseline characteristics were quite similar between treatment arms in the phase 2 portion of the clinical trial. The median age was 58 years with 42 patients ≥65 years of age. 86.4% of the patients were Caucasian. More than 25 different soft tissue sarcoma subtypes were represented in this trial, the most frequent being leiomyosarcoma (38.4%), undifferentiated pleomorphic sarcoma (18.1%) and liposarcoma (17.3%). Other subtypes were infrequently represented. Patients had received 0-4 previous lines of therapy for treatment of advanced disease but had not previously received treatment with anthracyclines. The number of patients receiving post-study systemic therapy was similar between arms. Ten patients in the olaratumab plus doxorubicin arm and 5 patients in the doxorubicin arm received post-study radiotherapy only. 3 patients in the olaratumab plus doxorubicin arm and 1 patient in the doxorubicin arm had post-study surgery only. 2 patients in the olaratumab plus doxorubicin arm and none in the doxorubicin arm received both post-study radiotherapy and surgery.
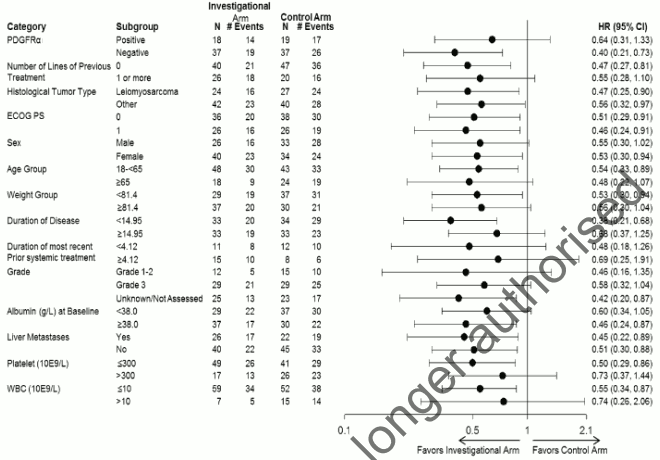
The median cumulative dose of doxorubicin was 487.6 mg/m² in the olaratumab plus doxorubicin arm and 299.6 mg/m² in the doxorubicin alone arm. The primary efficacy outcome measure was progression free survival (PFS) by investigator assessment. Key secondary efficacy outcome measures were overall survival (OS) and objective response rate (ORR) (see Table 3). The study met its primary endpoint (PFS). PFS according to a post-hoc, blinded, independent assessment was 8.2 months vs 4.4 months; HR=0.670; p=0.1208. A statistically significant improvement in OS was seen in the olaratumab plus doxorubicin arm in comparison to treatment with doxorubicin alone in the overall population. The main analysis was performed in the following two subgroups: Leiomyosarcoma (LMS) and non-LMS (other). Subgroups analysis of OS is shown in figure 2. Difference in objective response rate [complete response (CR) + partial response (PR)] according to investigator assessment was not statistically significant (18.2% vs 11.9% in patients randomised to olaratumab plus doxorubicin compared to patients randomized to doxorubicin respectively).
Efficacy results are shown in Table 3 and Figures 1 and 2.
Table 3. Summary of survival data - ITT population:
| Lartruvo plus doxorubicin (n=66) | Doxorubicin alone (n=67) | |
|---|---|---|
| Progression free survival, months* | ||
| Median (95% CI) | 6,6 (4,1, 8,3) | 4,1 (2,8, 5,4) |
| Hazard ratio (95% CI) | 0,672 (0,442, 1,021) | |
| p-value | 0,0615** | |
| Overall survival, months | ||
| Median (95% CI) | 26,5 (20,9, 31,7) | 14,7 (9,2, 17,1) |
| Hazard ratio (95% CI) | 0,463 (0,301, 0,710) | |
| p-value | 0,0003 | |
Abbreviations: CI = confidence interval
* By investigator assessment
** Met phase 2 protocol defined significance level of 0.19
Figure 1. Kaplan-Meier curves of overall survival for Lartruvo plus doxorubicin versus doxorubicin alone:
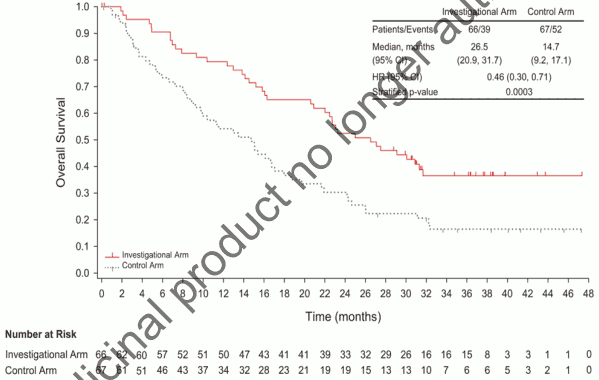
Figure 2. Forest plot for subgroup analysis of overall survival (ITT population):
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with olaratumab in one or more subsets of the paediatric population in soft tissue sarcoma (see section 4.2 for information on paediatric use).
This medicinal product has been authorised under a so-called 'conditional approval' scheme. This means that further evidence on this medicinal product is awaited. The European Medicines Agency will review new information on this medicinal product at least every year and this SmPC will be updated as necessary.
Pharmacokinetic properties
Absorption
Olaratumab is administered as an intravenous infusion only.
Distribution
The population pharmacokinetic (PopPK) model-based mean (CV%) volume of distribution of olaratumab at steady state (Vss) was 7.7 L (16%).
Elimination
The PopPK model-based mean (CV%) clearance for olaratumab was 0.56 L/day (33%). This corresponds to a mean terminal half-life of approximately 11 days.
Special populations
Age, sex, and race had no clinically meaningful effect on the PK of olaratumab based on a PopPK analysis. Clearance and volume of distribution had a positive correlation with body weight.
Renal impairment
No formal studies have been conducted to evaluate the effect of renal impairment on the PK of olaratumab. Based on a PopPK analysis, no clinically meaningful differences in the clearance of olaratumab were observed in patients with mild (calculated creatinine clearance [CLcr] 60-89 mL/min, n=43), or moderate (CLcr 30-59 mL/min, n=15) renal impairment compared to patients with normal renal function (CLcr ≥90 mL/min, n=85). No data were available from patients with severe renal impairment (CLcr 15-29 mL/min).
Hepatic impairment
No formal studies have been conducted to evaluate the effect of hepatic impairment on the PK of olaratumab. Based on a PopPK analysis, no clinically meaningful differences in the clearance of olaratumab were observed in patients with mild (total bilirubin within upper limit of normal [ULN] and AST>ULN, or total bilirubin >1.0-1.5 times ULN and any AST level, n=16), or moderate (total bilirubin >1.5-3.0 times ULN, n=1) hepatic impairment compared to patients with normal hepatic function (total bilirubin and AST ≤ ULN, n=126). No data were available from patients with severe hepatic impairment (total bilirubin >3.0 times ULN and any AST level).
Preclinical safety data
Non-clinical data reveal no special hazard for humans based on repeat dose toxicity studies in monkeys.
No animal studies have been performed to test olaratumab for potential of carcinogenicity, genotoxicity, or fertility impairment. Administration of an anti-murine PDGFR-α surrogate antibody to pregnant mice during organogenesis at 50 and 150 mg/kg resulted in increased malformations (abnormal eyelid development) and skeletal alterations (frontal/parietal additional ossification site). The foetal effects in mice administered the surrogate antibody occurred at exposures less than the AUC exposure at the maximum recommended human dose of 15 mg/kg olaratumab.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.