LAZCLUZE Film-coated tablet Ref.[114943] Active ingredients: Lazertinib
Source: European Medicines Agency (EU) Revision Year: 2025 Publisher: Janssen-Cilag International NV, Turnhoutseweg 30, B-2340 Beerse, Belgium
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: antineoplastic agents, protein kinase inhibitors
ATC code: L01EB09
Mechanism of action
Lazertinib is an irreversible EGFR tyrosine kinase inhibitor (TKI). It selectively inhibits both primary activating EGFR mutations (exon 19 deletions and exon 21 L858R substitution mutations) and the EGFR T790M resistance mutation, while having less activity against wild-type EGFR.
Pharmacodynamic effects
Based on the exposure-response analyses for safety, paraesthesia and stomatitis appeared to show a trend of increasing occurrence with increase in lazertinib exposure.
Cardiac electrophysiology
The QTc interval prolongation potential of lazertinib was evaluated by exposure-response (E-R) analysis conducted with clinical data from 243 NSCLC patients who received 20, 40, 80, 120, 160, 240 or 320 mg lazertinib once daily in a phase 1/II study. The E-R analysis revealed no clinically relevant relationship between lazertinib plasma concentration and change in QTc interval.
Clinical efficacy and safety
MARIPOSA is a randomised, open-label, active-controlled, multicentre phase 3 study assessing the efficacy and safety of Lazcluze in combination with amivantamab as compared to osimertinib monotherapy in the first-line treatment of patients with EGFR-mutated locally advanced or metastatic NSCLC not amenable to curative therapy. Patient samples were required to have one of the two common EGFR mutations (exon 19 deletion or exon 21 L858R substitution mutation), as identified by local testing. Tumour tissue (94%) and/or plasma (6%) samples for all patients were tested locally to determine EGFR exon 19 deletion and/or exon 21 L858R substitution mutation status using polymerase chain reaction (PCR) in 65% and next generation sequencing (NGS) in 35% of patients.
A total of 1074 patients were randomised (2:2:1) to receive Lazcluze in combination with amivantamab, osimertinib monotherapy, or Lazcluze monotherapy until disease progression or unacceptable toxicity. Lazcluze was administered at 240 mg orally once daily. Amivantamab was administered intravenously at 1050 mg (for patients <80 kg) or 1400 mg (for patients ≥80 kg) once weekly for 4 weeks, then every 2 weeks thereafter starting at week 5. Osimertinib was administered at a dose of 80 mg orally once daily. Randomisation was stratified by EGFR mutation type (exon 19 deletion or exon 21 L858R substitution mutation), race (Asian or non-Asian), and history of brain metastasis (yes or no).
Baseline demographics and disease characteristics were balanced across the treatment arms. The median age was 63 (range: 25–88) years with 45% of patients ≥65 years and 11% ≥75 years; 62% were female; and 59% were Asian, and 38% were White. Baseline Eastern Cooperative Oncology Group (ECOG) performance status was 0 (34%) or 1 (66%); 69% never smoked; 41% had prior brain metastases; and 90% had Stage IV cancer at initial diagnosis. With regard to EGFR mutation status, 60% were exon 19 deletions and 40% were exon 21 L858R substitution mutations.
Lazcluze in combination with amivantamab demonstrated a statistically significant improvement in progression-free survival (PFS) by BICR assessment.
Table 4, Figure 1 and Figure 2 summarise efficacy results for Lazcluze in combination with amivantamab.
Table 4. Efficacy results in MARIPOSA:
| Lazcluze + amivantamab (N=429) | Osimertinib (N=429) | |
|---|---|---|
| Progression-free survival (PFS)a | ||
| Number of events | 192 (45%) | 252 (59%) |
| Median, months (95% CI) | 23.7 (19.1, 27.7) | 16.6 (14.8, 18.5) |
| HR (95% CI); p-value | 0.70 (0.58, 0.85); p=0.0002 | |
| Overall survival (OS) | ||
| Number of events | 142 (33%) | 177 (41%) |
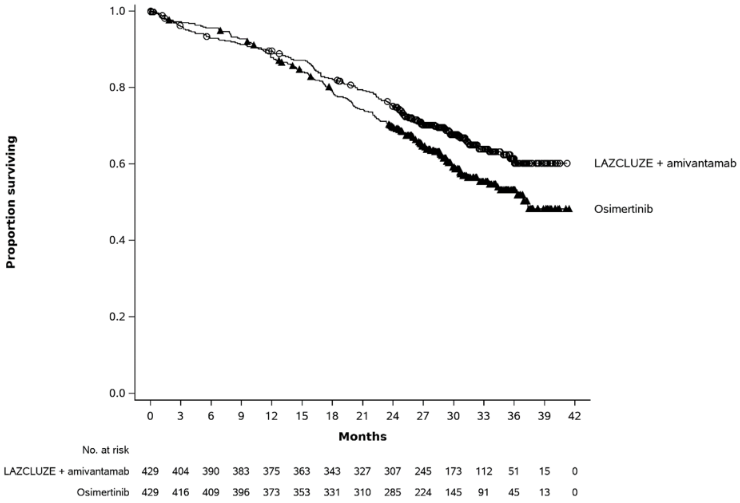
| Median, months (95% CI) | NE (NE, NE) | 37.3 (32.5, NE) |
| HR (95% CI); p-valueb | 0.77 (0.61, 0.96); p=0.0185 | |
| Objective response rate (ORR)a,c | ||
| ORR % (95% CI) | 80% (76%, 84%) | 77% (72%, 81%) |
| Duration of response (DOR)a,c | ||
| Median, months (95% CI) | 25.8 (20.3, 33.9) | 18.1 (14.8, 20.1) |
BICR = blinded independent central review; CI = confidence interval; NE = not estimable.
PFS results are from data cut-off 11 August 2023 with median follow-up of 22.0 months. OS, ORR, and DOR results are from data cut-off 13 May 2024 with median follow-up of 31.3 months.
a BICR by RECIST v1.1.
b The p-value is compared to a 2-sided significance level of 0.00001. Thus the OS results are not statistically significant as of the latest interim analysis.
c Based on confirmed responders.
Figure 1. Kaplan-Meier curve of PFS in previously untreated NSCLC patients by BICR assessment:
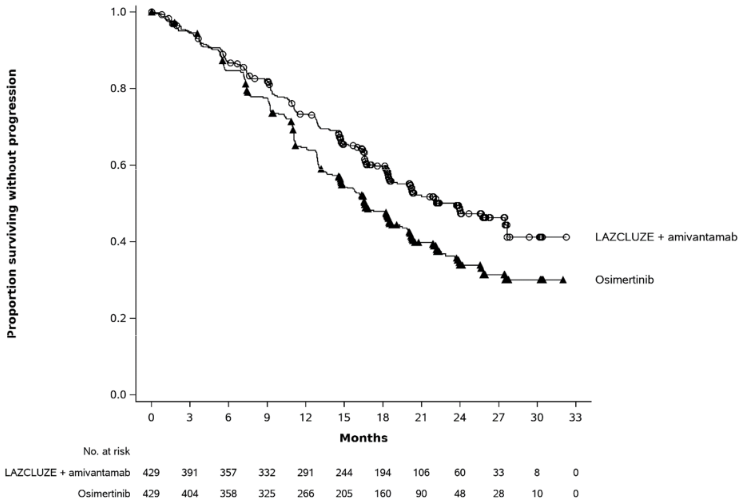
With a median follow up of approximately 31 months, the updated OS HR was 0.77 (95% CI: 0.61, 0.96; p=0.0185). This was not statistically significant as compared to a 2-sided significance level of 0.00001.
Figure 2. Kaplan-Meier curve of OS in previously untreated NSCLC patients:
Intracranial ORR and DOR by BICR were pre-specified endpoints in MARIPOSA. In the subset of patients with intracranial lesions at baseline, the combination of Lazcluze and amivantamab demonstrated similar intracranial ORR to the control. Per protocol, all patients in MARIPOSA had serial brain MRIs to assess intracranial response and duration. Results are summarised in Table 5.
Table 5. Intracranial ORR and DOR by BICR assessment in subjects with intracranial lesions at baseline:
| Lazcluze + amivantamab (N=180) | Osimertinib (N=186) | |
|---|---|---|
| Intracranial tumour response assessment | ||
| Intracranial ORR (CR+PR), % (95% CI) | 77% (70%, 83%) | 77% (71%, 83%) |
| Complete response | 63% | 59% |
| Intracranial DOR | ||
| Median, months (95% CI) | NE (21.4, NE) | 24.4 (22.1, 31.2) |
CI = confidence interval; NE = not estimable
Intracranial ORR and DOR results are from data cut-off 13 May 2024 with median follow-up of 31.3 months.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with Lazcluze in all subsets of the paediatric population in non-small cell lung cancer.
5.2. Pharmacokinetic properties
Following single and multiple once daily oral administration, lazertinib maximum plasma concentration (Cmax) and area under plasma concentration time curve (AUC) increased approximately dose proportionally across 20 to 320 mg dose range.
The steady state plasma exposure was achieved by day 15 of once daily administration and approximately 2-fold accumulation was observed at steady state with 240 mg once daily dose.
The lazertinib plasma exposure was comparable when lazertinib was administered either in combination with amivantamab or as a monotherapy.
Absorption
The median time to reach single dose and steady state Cmax was comparable and ranged from 2 to 4 hours.
Following administration of 240 mg lazertinib with a high-fat meal (800~1000 kcal, fat content approximately 50%), the Cmax and AUC of lazertinib were comparable to that under fasting conditions suggesting lazertinib can be taken with or without food.
Distribution
Lazertinib was extensively distributed, with mean (CV%) apparent volume of distribution of 4264 (43.2%) L at 240 mg dose. Lazertinib mean (CV%) plasma protein binding was approximately 99.2% (0.13%) in humans. Lazertinib demonstrated covalent binding to human blood and plasma proteins after oral dosing, and during in vitro incubations.
Metabolism
Lazertinib is primarily metabolised by glutathione conjugation, either enzymatic via glutathione-S-transferase (GST) or non-enzymatic, as well as by CYP3A4. The most abundant metabolites are glutathione catabolites and considered clinically inactive. The plasma exposure of lazertinib was affected by GSTM1 mediated metabolism, leading to lower exposure (less than 2-fold difference) in Non-null GSTM1 patients. No dose adjustment is required based on GSTM1 status.
Elimination
The mean (CV%) apparent clearance and terminal half-life of lazertinib at 240 mg dose were 44.5 (29.5%) L/h and 64.7 (32.8%) hours, respectively.
Excretion
Following a single oral dose of radiolabelled lazertinib, approximately 86% of the dose was recovered in faeces (<5% as unchanged) and 4% in urine (< 0.5% as unchanged).
Co-administration with OCT1 and UGT1A1 substrates
The co-administration of multiple doses of Lazcluze did not increase metformin (OCT1 substrate) Cmax and AUC. Lazcluze does not inhibit OCT1.
Based on in vitro studies, Lazcluze may inhibit UGT1A1. However, due to lack of effect on indirect bilirubin levels in clinical study, no clinically relevant interaction is expected with UGT1A1 substrates.
Special populations
Elderly
Based on population PK analysis, no clinically meaningful age-based differences in pharmacokinetics of lazertinib were observed.
Renal impairment
Based on population PK analysis, no dose adjustment is required for patients with mild, moderate or severe renal impairment with estimated glomerular filtration rate (eGFR) of 15 to 89 mL/min. Data in patients with severe renal impairment (eGFR of 15 to 29 mL/min) are limited (n=3), but there is no evidence to suggest that dose adjustment is required in these patients. No data are available in patients with end stage renal disease (eGFR <15 mL/min).
Hepatic impairment
Based on findings from clinical pharmacology study, moderate hepatic impairment (Child-Pugh Class B) had no clinically meaningful effect on lazertinib single dose PK. Based on population PK analysis, no dose adjustment is required for patients with mild (total bilirubin ≤ ULN and AST > ULN or ULN < total bilirubin ≤ 1.5×ULN and any AST) or moderate (1.5×ULN < total bilirubin ≤ 3×ULN and any AST) hepatic impairment. No data are available in patients with severe hepatic impairment (total bilirubin > 3×ULN and any AST).
Paediatric population
The pharmacokinetics of lazertinib in paediatric patients have not been investigated.
Other populations
No clinically meaningful differences in lazertinib PK were observed based on sex, body weight, race, ethnicity, baseline laboratory assessments (creatinine clearance, albumin, alanine aminotransferase, alkaline phosphatase, aspartate aminotransferase), ECOG performance status, EGFR mutation type, initial diagnosis cancer stage, prior therapies, brain metastasis, and history of smoking.
5.3. Preclinical safety data
The main findings observed in repeat-dose toxicity studies with lazertinib in rats and dogs comprised mild epithelial atrophy to degenerative erosions, inflammation, and necrosis affecting the eye (corneal atrophy) skin (thin and rough hair coat, hair follicle degeneration, alopecia, ulcer), liver (increased liver enzymes, Kupffer cell hypertrophy and hepatocellular necrosis), lungs (alveolar macrophage infiltrate, lung inflammation and hyperplasia of alveolar type II cells), kidney (tubular dilatation, papillary necrosis, higher urea nitrogen, creatinine (females only), inorganic phosphorus, and potassium), GI (oesophageal epithelial atrophy, villus blunting/fusion in duodenum, and jejunum, liquid faeces), reproductive system (testis tubular degeneration, hypospermia, decreased oestrous cycles and corpora lutea, atrophy in uterus and vagina) These findings were observed in animals in exposures ranges of 0.9-3.4x than estimated exposures of patients administered with the recommended dose (240 mg) and were fully or partially resolved during the recovery phases. The heart was considered a target organ in dog alone and occurred at exposure levels 7x to that of exposure levels expected at the human recommended dose.
Carcinogenicity and mutagenicity
No evidence of genotoxicity for lazertinib was observed in in vitro bacterial mutagenicity, in vitro chromosomal aberration, and in vivo micronucleus tests in rats. Long-term animal studies have not been conducted to evaluate the carcinogenic potential of lazertinib.
Reproductive toxicology
Based on studies in animals, male and female fertility may be impaired by treatment with lazertinib. Degenerative changes were present in the testes of rats and dogs resulting in reduced luminal sperm in dogs following exposure to lazertinib for 1 month at clinically relevant exposure levels. Decreased numbers of corpora lutea were noted in the ovaries of rats exposed to lazertinib for ≥1 month at clinically relevant exposure levels. In a fertility and early embryonic development study in male and female rats, lazertinib induced a decrease in the number of oestrus cycles, an increase in post-implantation loss and decreased live litter size at or below the dose level that approximated the human clinical exposure at the recommended dose of 240 mg.
Developmental toxicity was observed in embryo-foetal development studies in rats and rabbits. In rats, decreases in foetal body weights in association with maternal toxicity were observed at a maternal exposure approximately 4 times higher than the human clinical exposure at 240 mg. In rabbits, an increased incidence of a foetal skull bone fusion (zygomatic arch fused to the maxillary process) was observed at maternal exposures well below the human clinical exposure at 240 mg.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.