LIBMELDY Dispersion for infusion Ref.[27971] Active ingredients: Atidarsagene autotemcel
Source: European Medicines Agency (EU) Revision Year: 2023 Publisher: Orchard Therapeutics (Netherlands) B.V., Basisweg 10, 1043 AP Amsterdam, The Netherlands
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Other haematological agents
ATC code: A16AB21
Mechanism of action
Libmeldy is an ex vivo genetically modified autologous CD34+ hematopoietic stem and progenitor cell (HSPC) gene therapy. Autologous CD34+ HSPCs are collected from patient bone marrow (BM) harvest or from mobilised peripheral blood (mPB) and transduced with a lentiviral vector (ARSA LVV), which inserts one or more copies of the human ARSA complementary deoxyribonucleic acid (cDNA) into the cell's genome, so that genetically modified cells become capable of expressing the functional ARSA enzyme. When administered to the patient following the administration of a myeloablative conditioning regimen, the genetically modified cells engraft and are able to repopulate the haematopoietic compartment. A subpopulation of the infused HSPCs and/or their myeloid progeny is able to migrate across the blood brain barrier to the brain and engraft as central nervous system (CNS) resident microglia and perivascular CNS macrophages as well as endoneural macrophages in the peripheral nervous system (PNS). These genetically modified cells can produce and secrete the functional ARSA enzyme, which can be taken up by surrounding cells, a process known as cross-correction, and used to break down, or prevent the build-up, of harmful sulfatides.
Following successful and stable engraftment in the patient, the effects of the product are expected to be persistent.
Pharmacodynamic effects
Durable and stable peripheral engraftment of genetically modified cells was observed from 1-month post Libmeldy administration in all evaluable patients. A persistent vector copy number (VCN) was also observed in CD34+ cells isolated from the bone marrow throughout the follow-up period. These biological findings demonstrate a sustained multilineage engraftment of gene-corrected cells, which is essential for supporting the long-term production of ARSA and resulting long-term clinical benefit.
At Year 1 post-treatment, the proportion of BM-derived colonies harbouring the LVV genome (%LV) in the overall treated population was 54.8% (range: 20.0% to 100%, [N=23]). The proportion of BM-derived colonies harbouring the LVV genome (% LV) at Year 5 was 45.0% (range: 18.8% to 90.6% [n=6, 4 Late infantile (LI) and 2 Early Juvenile (EJ)]), indicative of stable engraftment over time in the treated population.
Reconstitution of ARSA activity in the hematopoietic system was observed in all MLD patients treated, with a progressive reconstitution of ARSA levels in Peripheral Blood Mononuclear Cells (PBMCs) which reached values within the normal reference range by 3 months post-treatment and remained stable within or above the normal range throughout the duration of the follow-up (see Figure 1).
Figure 1. ARSA activity in PBMCs over time (geometric mean and 95% CIs), by disease subtype (integrated efficacy set; N=29):
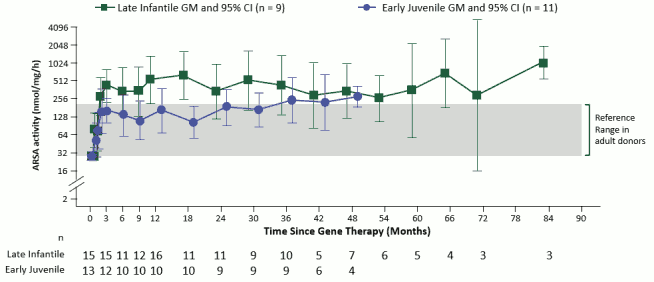
Note: Values < LLQ are imputed at LLQ. LLQ is 25.79 nmol/mg/h. GMs and 95% CIs are presented where there are at least 3 patients with non-missing data. ARSA: arylsulfatase A; CI: confidence interval; GM: geometric mean; LLQ: lower limit of quantification; PBMCs: peripheral blood mononuclear cells.
ARSA activity was also measured in cerebrospinal fluid (CSF) as a surrogate compartment of metabolic correction in the brain. The ARSA activity in CSF went from undetectable at Baseline to detectable in all evaluable patients by Month 6 post-treatment and reached reference range levels at Year 1 post-treatment. Thereafter, central reconstitution of ARSA enzymatic activity remained stable within the reference range.
Clinical efficacy
Clinical efficacy was based on the integrated analysis of results from 29 early-onset MLD patients treated with Libmeldy prepared as a fresh (non-cryopreserved) formulation. These results were generated in twenty (20) patients treated in the Registrational Study (Study 201222 - an open-label, non-randomized, single-arm safety and efficacy clinical trial) with a median duration of post-treatment follow-up of 4.0 years (range: 0.6 to 7.5 years) and nine (9) patients treated in the context of 3 expanded access programs with a median follow-up of 1.5 years (range: 0.99 years to 2.72 years). In addition, initial results from 9 patients treated in a further study with the commercial (cryopreserved) formulation of Libmeldy (Study 205756) are summarised below.
The MLD disease spectrum can present in a variety of clinical forms, primarily based on the age of onset of the first symptoms of the disease. Pre-symptomatic Late Infantile (LI) or Early Juvenile (EJ) MLD patients and early symptomatic EJ MLD patients with biallelic mutations in the ARSA gene leading to a reduction of the ARSA enzymatic activity were included in the clinical development of Libmeldy. 'Biallelic mutations leading to a reduction of the ARSA enzymatic activity' refers to mutations leading to partial or total disruption of the ARSA enzymatic activity and resulting in accumulation of sulfatides. These biallelic mutations exclude common neutral mutations described in association with ARSA pseudo-deficiency alleles.
Patients and disease characteristics
The MLD forms (variants) were defined by the presence of the following criteria during the clinical development:
- Late infantile (LI): age at onset of symptoms in the older sibling(s) ≤30 months and/or 2 null (0) mutant ARSA alleles and/or peripheral neuropathy at electroneurography (ENG) study.
- Early juvenile (EJ): age at onset of symptoms (in the patient or in the older sibling) between 30 months and before 7 years, and/or 1 null (0) and 1 residual (R) mutant ARSA allele(s) and/or peripheral neuropathy at ENG study. In the above definition, null (0) or residual (R) alleles refer to either known or novel mutations.
The symptomatic status of the patients was defined as follows:
Pre-symptomatic: at time of inclusion into the clinical studies, LI or EJ patients were without neurological impairment (disease-related symptoms), with or without signs of the disease revealed by instrumental evaluations i.e. electroneurographic study (ENG) and brain magnetic resonance imaging (MRI).
Based on an analysis of the baseline characteristics of pre-symptomatic LI and EJ patients treated during the clinical development program, the definition of pre-symptomatic status was further refined to maximise the treatment benefit. Taking the results of this analysis into account, treatment with Libmeldy of a pre-symptomatic patient should be considered:
- For a patient with the LI form of the disease, in the absence of a delay in achievement of independent standing, or a delay in achievement of independent walking, associated with abnormal signs at neurological evaluation.
- For a patient with the EJ form of the disease, in the absence of neurological signs or symptoms of the disease resulting in cognitive, motor, or behavioural functional impairment or regression (substantiated by neurological examination, gross motor function evaluation and/or age appropriate neuropsychological tests).
Early symptomatic: at time of inclusion into the clinical studies, early symptomatic EJ patients met the following 2 criteria: intelligence quotient (IQ) ≥70 and the ability to walk independently for ≥10 steps.
Based on the analysis of clinically relevant benefits on the motor and cognitive functions, efficacy was only demonstrated in patients treated before the onset of cognitive deterioration at a time when they were still able to walk independently. Taking these results into consideration, treatment with Libmeldy of a patient with an earlysymptomatic EJ form of the disease should be considered:
- If this patient is able to walk independently, which means that the patient's GMFC-MLD score is ≤1, and
- If the patient's cognitive function has not started declining, which means that the patient's IQ is ≥85.
At time of inclusion in the clinical studies, out of the 29 early-onset MLD patients, 20 were presymptomatic and 9 were early symptomatic, 16 had a diagnosis of LI MLD and 13 had a diagnosis of EJ MLD. All LI study patients and some EJ patients were identified after an older sibling had developed symptoms and received an MLD diagnosis, prompting testing in other family members.
Table 3. Summary of demographic characteristics by symptomatic status at time of gene therapy and by disease subtype (Integrated efficacy set):
| Pre-symptomatic patients | Early symptomatic patients | |||
|---|---|---|---|---|
| Late Infantile subgroup (N=15) | Early Juvenile subgroup (N=5) | Late Infantile subgroup (N=1) | Early Juvenile subgroup (N=8) | |
| Sex, n (%) | ||||
| Female | 5 (33) | 2 (40) | 1 (100) | 5 (63) |
| Male | 10 (67) | 3 (60) | 0 | 3 (38) |
| Age at GT, in months | ||||
| Median | 13.1 | 48.9 | 23.3 | 77.9 |
| Min | 7.6 | 11.4 | 23.3 | 38.8 |
| Max | 17.8 | 66.8 | 23.3 | 139.9 |
Mobilisation and apheresis
During the clinical development, all (ten) patients for whom the decision was made to use mPB as the source material – and not to conduct a BM harvest - were administered G-CSF (10-12.5 μg/kg/day) to mobilise CD34+ cells prior to the apheresis procedure. Starting from day 3 of G-CSF administration, an additional mobilising agent, plerixafor, was given once daily (0.24 mg/kg, subcutaneous) if clinically indicated depending on the white blood cells and CD34+ cell count in the patient's peripheral blood. Apheresis was performed as soon as the CD34+ cell count reached an adequate level, according to standard procedures.
If the target number of collected CD34+ cells to manufacture Libmeldy and to provide the back-up transplant were not reached with a single apheresis, a second procedure was performed. For all patients, the minimum number of CD34+ cells to manufacture Libmeldy (8 x 106 CD34+ cells/kg) was collected with 1 cycle of mobilisation and 1 or 2 apheresis.
Pre-treatment conditioning
All patients received systemic conditioning with busulfan prior to treatment with Libmeldy.
Thirteen patients (45%) were treated with a sub-myeloablative conditioning (SMAC) regimen, defined as a target cumulative AUC of 67,200 μg*h/L. Sixteen patients (55%) were treated with a myeloablative (MAC) conditioning regimen, defined as a target cumulative AUC of 85,000 μg*h/L.
For the SMAC conditioning regimen, patients received a total of 14 doses of busulfan (according to patient's weight), as a 2-hour IV infusion administered every 6 hours from Day -4 to Day -1. Busulfan plasma levels were monitored by serial pharmacokinetic sampling and adjusted using a target dose AUC of 4800 μg*h/L (range: 4200 to 5600 μg*h/L), which corresponds to an expected total cumulative AUC of 67,200 µg*h/L (range 58,800 to 78,400 μg*h/L). The average, cumulative AUC in patients who received a SMAC regimen was higher than expected but remained within the target range (geometric mean 71,923.53 [95% CI: 68,751.04, 75,242.41]).
For the MAC conditioning regimen, patients received body-surface area-based dosing of busulfan according to the patients age (80 mg/m²/dose if ≤1 year; 120 mg/m²/dose if >1 year) for a total of 4 doses, administered as a 3 hour IV infusion every 20 to 24 hours from Day -4 to Day -1. Busulfan plasma levels were monitored by serial pharmacokinetic sampling and adjusted using a target total cumulative AUC of 85,000 µg*h/L (range: 76,500 to 93,500 µg*h/L).
Subgroup analyses by conditioning regimen i.e. comparison of the subgroups of patients who received the MAC vs. the SMAC regimen, didn't show noticeable differences in the level of transduced cell engraftment nor in ARSA enzyme activity (in total PBMCs and BM-derived mononuclear cells). Moreover, the safety profiles of both regimens were shown to be comparable.
Therefore, the decision to use the MAC or SMAC regimen for pre-treatment conditioning is at the discretion of the treating physician, taking into consideration the patient's clinical characteristics such as, but not limited to, age, hepatic function, prematurity and thrombophilia.
During clinical development, prophylaxis for veno-occlusive disease (VOD) and related endothelial injury complications was required per institutional practice with ursodeoxycholic acid or defibrotide.
Libmeldy administration
All patients (N=29) were administered the medicinal product with a mean (min, max) cell dose of 10.81 x 106 (4.2, 25.9) CD34+ cells/kg as an intravenous infusion.
Integrated efficacy results (N=29)
The co-primary efficacy endpoints were:
- Gross Motor Function Measure (GMFM): An improvement of >10% of the total GMFM score in treated patients, when compared to the GMFM scores in the age-matched, untreated historical control MLD population (i.e., TIGET natural history [NHx] Study), evaluated at Year 2 after treatment (see Table 5), and
- ARSA activity: A significant (≥2 SD) increase in residual ARSA activity as compared to pretreatment values, measured in peripheral blood mononuclear cells (PBMC) at Year 2 after treatment (see Pharmacodynamic Effects, Figure 1 and Table 6).
Early-onset MLD patients treated before the onset of overt symptoms showed normal motor development, stabilisation, or delay in the rate of progression of motor dysfunction as measured by GMFM total score (%) (see Table 5).
Using an ANCOVA model adjusted for age at GMFM assessment and treatment, the mean difference between treated pre-symptomatic LI patients and age matched untreated LI patients from the NHx study was 71.0% at Year 2 and 79.8% at Year 3. Similarly, the mean difference between treated presymptomatic EJ patients and aged matched untreated EJ patients was 52.4% at Year 2 and 74.9% at Year 3. These treatment differences were statistically significant (p≤0.008) in favour of Libmeldy. Although not statistically significant, a clear difference in GMFM total score was also noted between treated early symptomatic EJ patients and aged matched untreated EJ patients (28.7% at Year 2; p=0.350 and 43.9% at Year 3; p=0.054).
Table 5. GMFM total score (%) at Year 2 and Year 3 in pre-symptomatic and earlysymptomatic patients (late infantile and early juvenile subgroups) with comparison to age-matched natural history data (integrated efficacy set):
| Adjusted mean GMFM total score | Mean treatment difference in GMFM total score between treated patients and age- matched untreated natural history patients | |||
|---|---|---|---|---|
| Treated patients | Untreated natural history patients | |||
| Pre-symptomatic patients | Late infantile | |||
| Year 2* | 79.5% (n=10) | 8.4% (n=8) | 71.0% (95% CI: 60.4-81.7); p<0.001 | |
| Year 3M | 82.6% (n=9) | 2.8% (n=9) | 79.8% (95% CI: 66.2-93.3); p<0.001 | |
| Early juvenile | ||||
| Year 2* | 96.7% (n=4) | 44.3% (n=8) | 52.4% (95% CI: 25.1-79.6); p=0.008 | |
| Year 3 | 93.2% (n=4) | 18.2% (n=9) | 74.9% (95% CI: 50.8-99.1); p<0.001 | |
| Early-Symptomatic patients | Early juvenile | |||
| Year 2* | 60.7% (n=6) | 31.9% (n=10) | 28.7% (95% CI: -14.1-71.5); p=0.350 | |
| Year 3 | 59.8% (n=6) | 15.9% (n=10) | 43.9% (95% CI: 9.2-78.5); p=0.054 | |
* The Gross Motor Function Measure at two years after treatment was a co-primary endpoint of the registrational clinical study. Note: Analysis of covariance adjusting for treatment and age. P-values are from a two-sided 5% hypothesis test with null hypothesis of 10% difference. CI: confidence interval; EJ: early juvenile; GMFM: gross motor function measurement; LI: late infantile; MLD: metachromatic leukodystrophy.
Deterioration of gross motor function was assessed from disease onset in EJ patients who were earlysymptomatic at the time of gene therapy. By four years post disease onset, the estimated proportion of patients who survived and maintained locomotion and ability to sit without support (GMFC-MLD level 5 or higher) was 62.5% in the treated group compared to 26.3% in the untreated group, representing a delay in disease progression following treatment with Libmeldy.
A statistically significant increase in ARSA activity in PBMCs was also observed at Year 2 posttreatment compared to pre-treatment baseline in both pre-symptomatic patients (20.0-fold increase; p<0.001) and early symptomatic patients (4.2-fold increase; p=0.004) (See Table 5).
Table 6. ARSA activity measured in PBMCs (geometric mean) at Baseline and Year 2 after treatment in pre-symptomatic and early-symptomatic patients (integrated efficacy set):
| Geometric mean (%CVb) ARSA Activity in PBMCs | Fold Increase from Baseline to Year 2* | ||
|---|---|---|---|
| Baseline | Year 2 | ||
| Pre-symptomatic | 26.923 (16.72) (n=19) | 339.736 (270.85) (n=14) | 20.0 (95% CI: 9.0, 44.0) p<0.001 |
| Early-symptomatic | 26.025 (2.72) (n=9) | 134.056 (55.94) (n=6) | 4.2 (95% CI: 1.6, 11.2) p=0.004 |
* Ratio in adjusted means from a mixed model repeated measures of data on the log scale, adjusting for visit, baseline, baseline*visit, disease subtype and disease subtype*visit
A secondary efficacy endpoint of the integrated efficacy analysis was measurement of IQ above 55 post-treatment with Libmeldy, the threshold for moderate mental retardation (DSM-IV), using neuropsychological tests. Intelligence Quotient/Development Quotient (IQ/DQ) measures, i.e. cognitive and language abilities, complement results from the GMFM and provide further evidence that the high levels of engraftment and enzymatic reconstitution translate into relevant treatment effects on key symptomatic domains in MLD patients.
In the LI subgroup (all pre-symptomatic at time of treatment except one), 12 out of 15 assessed patients had a fairly constant IQ/DQ, within the normal range (IQ/DQ score of 100 +/- SD of 15) throughout follow-up. All but 2 of these patients (one pre-symptomatic, one early-symptomatic) remained above the threshold of severe mental disability (IQ/DQ >55) at chronological ages at which all 14 untreated NHx patients with neuropsychological assessments showed evidence of severe cognitive impairment (i.e. IQ/DQ below 55 and close to 0).
Of the 10 surviving EJ patients, all 4 pre-symptomatic patients and 4 out of 6 early-symptomatic patients showed normal IQ/DQ throughout follow-up. In contrast, 11 out of 12 NHx patients with neuropsychological assessments showed evidence of severe cognitive impairment during follow-up.
At the time of the integrated data analysis, i.e. at a median follow-up time of 3.035 years posttreatment (range 0.99 to 7.51), none of the 16 patients in the treated LI subgroup, all pre-symptomatic at time of treatment except one, had died (100% overall survival). Four pre-symptomatic LI patients were alive 6 or more years after treatment and 2 pre-symptomatic LI patients were alive 7 or more years after treatment. In comparison, 12 out of 19 (63.2%) untreated LI patients in the NHx study had died at the time of the analysis.
Comparable overall survival was observed in the treated and untreated EJ groups with a median follow-up time of 3.49 years post-treatment (range 0.64 to 6.55). One out of 5 (20%) EJ patients treated at pre-symptomatic stage died, due to cerebral ischemic infarction, not deemed related to Libmeldy. There were 2 deaths among the 8 (25.0%) EJ patients treated at early-symptomatic stage, both due to disease progression, and also not considered to be related to Libmeldy treatment. Similarly, 3 of the 12 (25%) untreated EJ patients in the NHx study had died at the time of the analysis.
A sensitivity analysis conducted to identify clinical factors, which could have influenced the level of treatment benefit with Libmeldy and optimize the recommended use of the treatment, identified 4 treatment failures:
- One LI patients experienced onset of disease-related symptoms between screening and administration of Libmeldy and was considered symptomatic at the time of treatment. The progression of this patient post-treatment was comparable to untreated NHx patients in both cognitive function and motor development.
- Three early symptomatic EJ patients treated with Libmeldy showed deterioration in both motor and cognitive functions comparable to that observed in untreated NHx patients and progression of the disease led to death in two of them. Two out of the three patients showed IQ <85 (82 and 58) at the time of treatment. Two out of the three patients showed deterioration between screening and baseline (onset of conditioning regimen) assessments.
Study 205756 (cryopreserved commercial formulation)
Study 205756 is an open-label, single-arm study to evaluate the cryopreserved (commercial) formulation of Libmeldy in the treatment of pre-symptomatic LI and pre-symptomatic and early symptomatic EJ MLD patients. The cell dose range used in the first 9 patients in Study 205756 (10.45-30.0 x 106 CD34+ cells/kg) is close to the range used in patients treated with the fresh (investigational) formulation of the medicinal product (4.2-25.9 x 106 CD34+ cells/kg).
At the time of data cut, 6 patients (3LIs, 3EJs), all pre-symptomatic at the time of treatment, have been treated, with a median follow-up post-treatment of 0.87 year (range: 0.0 to 1.47 years). Preliminary efficacy data show levels of engraftment, Vector Copy Number, ARSA activity in PBMCs and CSF at different timepoints post-gene therapy within the range observed in the integrated data analysis of the patients treated with the fresh formulation of Libmeldy.
Preliminary safety data indicate that Libmeldy was well tolerated. The safety profile observed in this study with the cryopreserved formulation is consistent with the profile established in patients treated with the fresh formulation in terms of nature, time of onset and frequency of reported adverse events.
Paediatric population
Libmeldy has been studied in infants and children with an age range between 7.6 months and 11.6 years.
The European Medicines Agency has deferred the obligation to submit the results of studies with Libmeldy in the late juvenile subset of the paediatric population with metachromatic leukodystrophy (i.e. MLD patients aged between 7 and less than 17 years at time of disease onset) (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Libmeldy is a gene therapy medicinal product consisting of autologous cells that have been genetically modified ex vivo. The nature of Libmeldy is such that conventional studies on pharmacokinetics, absorption, metabolism, and elimination are not applicable. The biodistribution of Libmeldy was nonetheless studied and distribution to haematopoietic tissues and disease target organs (including the brain) was demonstrated.
5.3. Preclinical safety data
Due to the nature of Libmeldy, a standard toxicological assessment was not applicable and conventional mutagenicity, carcinogenicity and reproductive and developmental toxicity studies have not been conducted.
The pharmacology, toxicology and genotoxicity of Libmeldy were evaluated in vitro and in vivo. Integration site analysis (ISA) of mouse Lin- bone marrow cells and human CD34+ cells transduced with ARSA LVV was conducted pre- and post-transplantation into mice and showed no enrichment for insertion in or near cancer-related genes, or clonal dominance. A prototype lentiviral vector related to ARSA LVV did not induce in vitro transformation and sustained growth of transduced wild type mouse Lin-bone marrow cells due to insertional transformation. Lin- bone marrow cells from Cdkn2a-/- mice, a strain prone to cancer triggered by gamma-retroviral insertional mutagenesis, transduced with the same prototype lentiviral vector did not show genotoxic potential when transplanted into wild type mice.
Toxicity and oncogenesis (tumorigenicity) studies were performed in the mouse model of MLD. No evidence of toxicity due to ARSA overexpression and no abnormal or malignant growth of transplanted cells or hematopoietic tumours related to the integration of ARSA LVV were observed. ARSA overexpression in human HSPCs and in ARSA Tg mice did not impair the activation of other sulfatases dependent on the sulfatase activator SUMF-1, did not affect the proliferation and differentiation capacities of transduced cells and did not induce toxicity or functional impairment in ARSA Tg mice.
Additional studies with human CD34+ cells transduced with ARSA LVV administered to immunodeficient, myeloablated mice demonstrated no toxicity, no vector mobilisation and bystander transduction of male gonads.
Molecular monitoring did not detect replication competent lentivirus (RCL).
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.