LOJUXTA Hard, capsule Ref.[27700] Active ingredients: Lomitapide
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: Chiesi Farmaceutici S.p.A., Via Palermo 26/A, 43122 Parma, Italy
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Lipid modifying agents, other lipid modifying agents
ATC code: C10AX12
Mechanism of action
Lomitapide is a selective inhibitor of microsomal transfer protein (MTP), an intracellular lipid-transfer protein that is found in the lumen of the endoplasmic reticulum and is responsible for binding and shuttling individual lipid molecules between membranes. MTP plays a key role in the assembly of apo B containing lipoproteins in the liver and intestines. Inhibition of MTP reduces lipoprotein secretion and circulating concentrations of lipoprotein-borne lipids including cholesterol and triglycerides.
Clinical efficacy and safety
A single arm, open-label study (UP1002/AEGR-733-005) evaluated the efficacy and safety of lomitapide when co-administered with a low-fat diet and other lipid-lowering therapies in adult patients with HoFH. Patients were instructed to maintain a low-fat diet (<20% calories from fat) and their lipid-lowering therapies at study entry, including apheresis if applicable, from 6 weeks prior to baseline through at least Week 26. The dose of lomitapide was escalated from 5 mg to an individually determined maximum tolerated dose up to 60 mg. After Week 26, patients remained on lomitapide to determine the effects of longer-term treatment and were allowed to change background lipid-lowering therapies. The study provided for a total of 78 weeks of treatment.
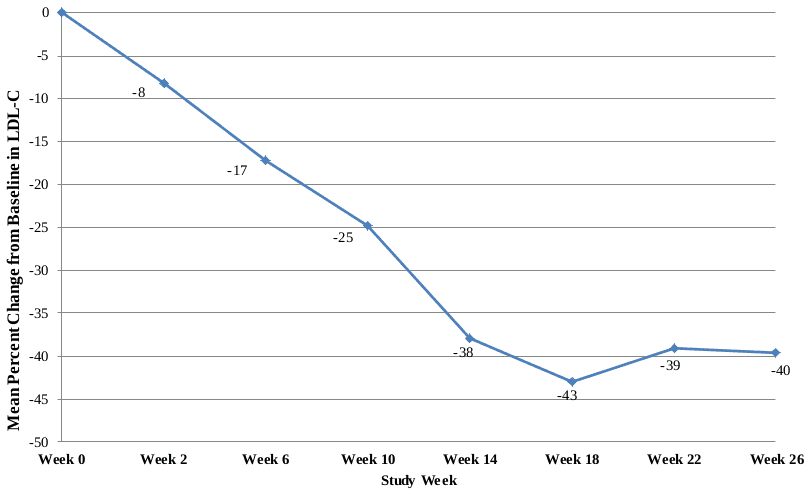
Twenty-nine patients were enrolled, of whom 23 completed through Week 78. Sixteen males (55%) and 13 females (45%) were included with a mean age of 30.7 years, ranging from 18 to 55 years. The mean dose of lomitapide was 45 mg at Week 26 and 40 mg at Week 78. At Week 26, the mean percent change in LDL-C from baseline of LDL-C was -40% (p<0.001) in the Intent to Treat (ITT) population. Mean percent change from baseline through Week 26 using last observation carried forward (LOCF) to each assessment is shown in Figure 1.
Figure 1. Mean percent changes from baseline in LDL-C in the major effectiveness study UP1002/AEGR-733-005 through Week 26 (the Primary Endpoint) using LOCF to each assessment (N=29):
Changes in lipids and lipoproteins through Week 26 and Week 78 of lomitapide treatment are presented in Table 5.
Table 5. Absolute values and percent changes from baseline to Weeks 26 and 78 in lipids and lipoproteins (major effectiveness study UP1002/AEGR-733-005):
| Parameter (units) | Baseline | Week 26/LOCF (N=29) | Week 78 (N=23) | ||||
|---|---|---|---|---|---|---|---|
| Mean (SD) | Mean (SD) | % Change | p-valueb | Mean (SD) | % Change | p-valueb | |
| LDL-C, direct (mg/dL) | 336 (114) | 190 (104) | -40 | <0.001 | 210 (132) | -38 | <0.001 |
| Total Cholesterol (TC) (mg/dL) | 430 (135) | 258 (118) | -36 | <0.001 | 281 (149) | -35 | <0.001 |
| Apolipoprotein B (apo B) (mg/dL) | 259 (80) | 148 (74) | -39 | <0.001 | 151 (89) | -43 | <0.001 |
| Triglycerides (TG) (mg/dL)a | 92 | 57 | -45 | 0.009 | 59 | -42 | 0.012 |
| Non high-density lipoprotein cholesterol (Non-HDL-C) (mg/dL) | 386 (132) | 217 (113) | -40 | <0.001 | 239 (146) | -39 | <0.001 |
| Very-low-density lipoprotein cholesterol (VLDL-C) (mg/dL) | 21 (10) | 13 (9) | -29 | 0.012 | 16 (15) | -31 | 0.013 |
| Lipoprotein (a) (Lp(a)) (nmol/L)a | 66 | 61 | -13 | 0.094 | 72 | -4 | <0.842 |
| High-density lipoprotein cholesterol (HDL-C) (mg/dL) | 44 (11) | 41 (13) | -7 | 0.072 | 43 (12) | -4.6 | 0.246 |
a Median presented for TG and Lp(a). p-value is based on the mean percent change
b p-value on the mean percent change from baseline based on paired t-test
At both Week 26 and Week 78, there were significant reductions in LDL-C, TC, apo B, TG, non-HDL-C, VLDL-C and changes in HDL-C trended lower at Week 26 and returned to baseline levels by Week 78.
The effect of Lojuxta on cardiovascular morbidity and mortality has not been determined.
At baseline, 93% were on a statin, 76% were on ezetimibe, 10% on niacin, 3% on a bile acid sequestrant and 62% were receiving apheresis. Fifteen of 23 (65%) patients had their lipid-lowering treatment reduced by Week 78, including planned and unplanned reductions/interruptions. Apheresis was discontinued in 3 out of 13 patients who were on it at Week 26, and frequency was reduced in 3 patients while maintaining low LDL-C levels through Week 78. The clinical benefit of reductions in background lipid-lowering therapy, including apheresis, is not certain.
Of the 23 patients who completed through Week 26, 19 (83%) had LDL-C reductions ≥25% with 8 (35%) having LDL-C <100 mg/dL and 1 having LDL-C <70 mg/dL at that time point.
In this study, 10 patients experienced elevations in AST and/or ALT >3 x ULN (see Table 6).
Table 6. Highest liver function test results post first dose (major effectiveness study UP1002/AEGR-733-005):
| Parameter/Abnormality | N (%) |
|---|---|
| ALT | |
| Number of Patients with Assessments | 29 |
| >3 to ≤5 x ULN | 6 (20.7) |
| >5 to ≤10 x ULN | 3 (10.3) |
| >10 to ≤20 x ULN | 1 (3.4) |
| >20 x ULN | 0 |
| AST | |
| Number of Patients with Assessments | 29 |
| >3 to ≤5 x ULN | 5 (17.2) |
| >5 to ≤10 x ULN | 1 (3.4) |
| >10 to ≤20 x ULN | 0 |
| >20 x ULN | 0 |
Elevations in ALT and/or AST >5 x ULN were managed with a dose reduction or temporary suspension of lomitapide dosing, and all patients were able to continue with study drug treatment. No clinically meaningful elevations in total bilirubin or alkaline phosphatase were observed. Hepatic fat was prospectively measured using MRS in all eligible patients during the clinical study (Table 7). Data from individuals who had repeat measurements after stopping lomitapide show that hepatic fat accumulation is reversible, but whether histological sequelae remain is unknown.
Table 7. Maximum categorical changes in % hepatic fat (major effectiveness study UP1002/AEGR-733-005):
| Maximum absolute increase in % hepatic fat | Efficacy phase weeks 0-26 N (%) | Safety phase weeks 26-78 N (%) | Entire trial weeks 0-78 N (%) |
|---|---|---|---|
| Number of evaluable patients | 22 | 22 | 23 |
| ≤5% | 9 (41) | 6 (27) | 5 (22) |
| >5% to ≤10% | 6 (27) | 8 (36) | 8 (35) |
| >10% to ≤15% | 4 (18) | 3 (14) | 4 (17) |
| >15% to ≤20% | 1 (5) | 4 (18) | 3 (13) |
| >20% to ≤25% | 1 (5) | 0 | 1 (4) |
| >25% | 1 (5) | 1 (5) | 2 (9) |
The European Medicines Agency has deferred the obligation to submit the results of studies with Lojuxta in one or more subsets of the paediatric population in HoFH (see section 4.2 for information on paediatric use).
This medicinal product has been authorised under 'exceptional circumstances'. This means that due to the rarity of the disease it has not been possible to obtain complete information on this medicinal product.
The European Medicines Agency will review any new information which may become available every year and this SmPC will be updated as necessary.
5.2. Pharmacokinetic properties
Absorption
The absolute oral bioavailability of lomitapide is 7%. Absorption is not limited by penetration of the active substance across the intestinal barrier but is predominantly influenced by an extensive first pass effect. Peak plasma concentrations of lomitapide were reached 4-8 hours following oral dosing. Lomitapide pharmacokinetics is approximately dose-proportional for oral single doses in the therapeutic range. Doses higher than 60 mg suggest a trend toward nonlinearity and are not recommended.
Upon multiple dosing Cmax and AUC increased in approximate proportion to lomitapide dose. Cmax and AUC were increased following either a high-fat meal (77% and 58%, respectively) or low fat meal (70% and 28%, respectively). Accumulation of lomitapide in plasma was consistent with that predicted after a single dose following once daily oral dosing above 25 mg for up to 4 weeks. Inter-individual variability in lomitapide AUC was approximately 50%.
At steady state the accumulation of lomitapide was 2.7 at 25 mg and 3.9 at 50 mg.
Distribution
Following intravenous administration, the volume of distribution of lomitapide was high (mean = 1 200 litres) despite a high degree (> 99.8%) of binding to plasma protein. In animal studies lomitapide was highly concentrated (200-fold) in the liver.
Biotransformation
Lomitapide is extensively metabolised, predominantly by CYP3A4. CYP isoforms 2E1, 1A2, 2B6, 2C8, and 2C19 are involved to a lesser extent and isoforms 2D6 and 2C9 are not involved in the metabolism of lomitapide.
Elimination
Following administration of a radiolabeled oral solution dose to healthy subjects, 93% of the administered dose was recovered in urine and faeces. Approximately 33% of the radioactivity was excreted in urine as metabolites. The remainder was excreted in faeces, primarily as oxidised metabolites. The elimination half-life of lomitapide was approximately 29 hours.
Special populations
Data in the pivotal clinical study were analysed with respect to the impact of potential covariates on lomitapide exposure. Of the parameters examined (race, body mass index (BMI), gender, weight, age), only BMI could be classified as a potential covariate.
Age and gender
There was no clinically relevant effect of age (18-64 years) or gender on the pharmacokinetics of lomitapide. Lomitapide has not been investigated in patients aged 65 years or older.
Race
No dose adjustment is required for Caucasian or Latino patients. There is insufficient information to determine if Lojuxta requires dose adjustment in other races. However, since the medicinal product is dosed in an escalating fashion according to individual patient safety and tolerability, no adjustment to the dosing regimen is recommended based on race.
Renal insufficiency
In the renal impairment population, lomitapide was only studied in patients with end-stage renal disease (ESRD). A pharmacokinetic study in patients with ESRD undergoing hemodialysis demonstrated a 36% increase in mean lomitapide plasma concentration compared to matched healthy controls. The terminal half-life of lomitapide was not affected.
Hepatic insufficiency
A single-dose, open-label study was conducted to evaluate the pharmacokinetics of 60 mg lomitapide in healthy volunteers with normal hepatic function compared with patients with mild (Child-Pugh A) and moderate (Child-Pugh B) hepatic impairment. In patients with moderate hepatic impairment, lomitapide AUC and Cmax were 164% and 361% higher, respectively, compared with healthy volunteers. In patients with mild hepatic impairment, lomitapide AUC and Cmax were 47% and 4% higher, respectively, compared with healthy volunteers. Lojuxta has not been studied in patients with severe hepatic impairment (Child-Pugh score 10-15).
Paediatric population
Lomitapide has not been investigated in children less than 18 years of age.
5.3. Preclinical safety data
In repeat-dose oral toxicology studies in rodents and dogs, the principal drug-related findings were lipid accumulation in the small intestine and/or liver associated with decreases in serum cholesterol and/or triglyceride levels. These changes are secondary to the mechanism of action of lomitapide. Other liver-related changes in repeat-dose toxicity studies in rats and dogs included increased serum aminotransferases, subacute inflammation (rats only), and single-cell necrosis. In a 1 year repeat-dose study in dogs there were no microscopic changes in the liver although serum AST was minimally increased in females.
Pulmonary histiocytosis was observed in rodents. Decreased red blood cell parameters as well as poikilocytosis and/or anisocytosis were observed in dogs. Testicular toxicity was observed in dogs at 205 times the human exposure (AUC) at 60 mg in a 6-month study. No adverse effects on the testes were observed in a 1-year study in dogs at 64 times the human exposure at 60 mg.
In a dietary carcinogenicity study in mice, lomitapide was administered up to 104 weeks at doses ranging from 0.3 to 45 mg/kg/day. There were statistically significant increases in the incidences of liver adenoma and carcinoma at doses ≥1.5 mg/kg/day in males (≥2 times the human exposure at 60 mg daily based on AUC) and ≥7.5 mg/kg/day in females (≥9 times the human exposure at 60 mg based on AUC). Incidences of small intestinal carcinoma and/or combined adenoma and carcinoma (rare tumours in mice) were significantly increased at doses ≥15 mg/kg/day in males (≥26 times the human exposure at 60 mg based on AUC) and at 15 mg/kg/day in females (22 times the human exposure at 60 mg based on AUC).
In an oral carcinogenicity study in rats, lomitapide was administered up to 99 weeks at doses up to 7.5 mg/kg/day in males and 2.0 mg/kg/day in females. Focal hepatic fibrosis was observed in males and females and hepatic cystic degeneration was observed in males only. In high-dose males, an increased incidence of pancreatic acinar cell adenoma was observed at an exposure 6 times that in humans at 60 mg based on AUC.
Lomitapide was not mutagenic or genotoxic in a battery of in vitro and in vivo studies.
Lomitapide had no effect on reproductive function in female rats at doses up to 1 mg/kg or in male rats at doses up to 5 mg/kg. Systemic exposures to lomitapide at these doses were estimated to be 4 times (females) and 5 times (males) higher than the human exposure at 60 mg based on AUC.
Lomitapide was teratogenic in rats in the absence of maternal toxicity at an exposure (AUC) estimated to be twice that in humans at 60 mg. There was no evidence of embryofoetal toxicity in rabbits at 3 times the maximum recommended human dose (MRHD) of 60 mg based on body surface area. Embryofoetal toxicity was observed in rabbits in the absence of maternal toxicity at ≥6.5 times the MRHD. In ferrets, lomitapide was both maternally toxic and teratogenic at <1 times the MRHD.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.