Source: European Medicines Agency (EU) Revision Year: 2020 Publisher: Novartis Europharm Limited, Vista Building, Elm Park, Merrion Road, Dublin 4, Ireland
Pharmacotherapeutic group: not yet assigned
ATC code: not yet assigned
The retinal pigment epithelium-specific 65 kilodalton protein (RPE65) is located in the retinal pigment epithelial cells and converts all-trans-retinol to 11-cis-retinol, which subsequently forms the chromophore, 11-cis-retinal, during the visual (retinoid) cycle. These steps are critical in the biological conversion of a photon of light into an electrical signal within the retina. Mutations in the RPE65 gene lead to reduced or absent RPE65 all-trans-retinyl isomerase activity, blocking the visual cycle and resulting in vision loss. Over time, accumulation of toxic precursors leads to the death of retinal pigment epithelial cells, and subsequently to progressive photoreceptor cell death. Individuals with biallelic RPE65 mutation-associated retinal dystrophy exhibit vision loss, including impaired visual function parameters such as visual acuity and visual fields often during childhood or adolescence; this loss of vision ultimately progresses to complete blindness.
Injection of voretigene neparvovec into the subretinal space results in transduction of retinal pigment epithelial cells with a cDNA encoding normal human RPE65 protein (gene augmentation therapy), providing the potential to restore the visual cycle.
The long-term safety and efficacy of Luxturna were assessed in a Phase 1 safety and dose escalation study (101), in which 12 subjects received unilateral subretinal injections of voretigene neparvovec; a follow-on study (102) in which voretigene neparvovec was administered to the contralateral eye in 11 of the 12 subjects who participated in the dose escalation study; a one-year, open-label Phase 3 controlled study (301) in which 31 subjects were randomised at two sites; and the continuation of the Phase 3 study, in which the 9 control subjects crossed over and received the intervention. A total of 41 subjects (81 eyes injected [one Phase 1 subject did not meet eligibility criteria for a second injection]) participated in the clinical programme. All participants had a clinical diagnosis of Leber congenital amaurosis, and some may have also had prior or additional clinical diagnoses, including retinitis pigmentosa. Confirmed biallelic RPE65 mutations and the presence of sufficient viable retinal cells (an area of retina within the posterior pole of >100 micron thickness, as estimated by optical coherence tomography [OCT]) were established for all participants.
Study 301 was an open-label, randomised, controlled study. 31 subjects were enrolled, 13 males and 18 females. The average age was 15 years (range 4 to 44 years), including 64% paediatric subjects (n=20, age from 4 to 17 years) and 36% adults (n=11). All subjects had a diagnosis of Leber’s congenital amaurosis owing to RPE65 mutations confirmed by genetic analysis in a certified laboratory.
21 subjects were randomised to receive subretinal injection of voretigene neparvovec. Visual acuity (LogMAR) of the first eye of these subjects at baseline was 1.18 (0.14), mean (SE). One subject discontinued from the study prior to treatment. 10 subjects were randomised to the control (non-intervention) group. Visual acuity (LogMAR) of the first eye of these subjects at baseline was 1.29 (0.21), mean (SE). One subject in the control group withdrew consent and was discontinued from the study. The nine subjects who were randomised to the control group were crossed over to receive subretinal injection of voretigene neparvovec after one year of observation. Each eye was administered a single subretinal injection of 1.5 × 1011 vg voretigene neparvovec in a total volume of 300 μL. The interval between injection to the eyes for each subject was from 6 to 18 days.
The primary endpoint of the Phase 3 study measured the mean change from baseline to one year in binocular multi-luminance mobility testing (MLMT) between the intervention and control groups. The MLMT was designed to measure changes in functional vision, specifically the ability of a subject to navigate a course accurately and at a reasonable pace at different levels of environmental illumination. This ability depends on the subject’s visual acuity, visual field and the extent of nyctalopia (decreased ability to perceive and/or see in dim light), each of which are functions specifically affected by the retinal disease associated with RPE65 mutations. In the Phase 3 study, the MLMT used seven levels of illumination ranging from 400 lux to 1 lux (corresponding to, for example, a brightly lit office down to a moonless summer night). The testing of each subject was videotaped and assessed by independent graders. A positive change score reflects passing the MLMT at a lower light level and a lux score of 6 reflects the maximum possible MLMT improvement. Three secondary endpoints were also tested: full-field light sensitivity threshold (FST) testing using white light; the change in MLMT score for the first assigned eye; and visual acuity (VA) testing.
At baseline, subjects achieved pass marks on the mobility test at between 4 and 400 ambient lux.
Table 4. Changes in MLMT score: year 1, compared to baseline (ITT population: n=21 intervention, n=10 control):
| Change in MLMT score | Difference (95% CI) Intervention-Control | p-value |
|---|---|---|
| using binocular vision | 1.6 (0.72, 2.41) | 0.001 |
| using assigned first eye only | 1.7 (0.89, 2.52) | 0.001 |
| using assigned second eye only | 2.0 (1.14, 2.85) | <0.001 |
The monocular MLMT change score significantly improved in the treatment group and was similar to the binocular MLMT results (see Table 4).
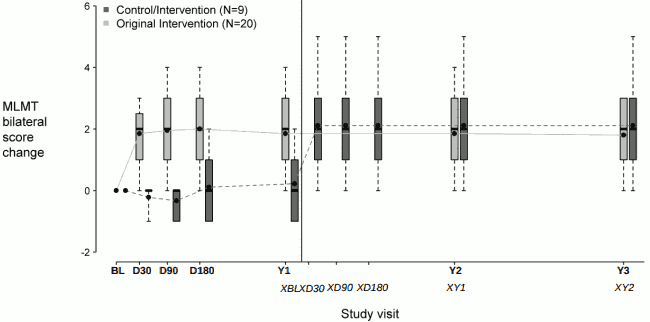
Figure 2 shows the effect of the medicinal product over the three-year period in the voretigene neparvovec treatment group, as well as the effect in the control group after crossing over to receive subretinal injection of voretigene neparvovec. Significant differences in binocular MLMT performance were observed for the voretigene neparvovec treatment group at day 30 and were maintained over the remaining follow-up visits throughout the three-year period, compared to no change in the control group. However, after crossing-over to receive subretinal injection of voretigene neparvovec, the subjects in the control group showed a similar response to the voretigene neparvovec as compared to the subjects in the voretigene neparvovec treatment group.
Figure 2. Change in MLMT score using binocular vision versus time before/after exposure to voretigene neparvovec:
Each box represents the middle 50% of distribution of MLMT score change. Vertical dotted lines represent additional 25% above and below the box. The horizontal bar within each box represents the median. The dot within each box represents the mean. The solid line connects the mean MLMT score changes over visits for the treatment group. The dotted line connects the mean MLMT score change over visits for the control group, including five visits during the first year without receiving voretigene neparvovec. The control group was administered voretigene neparvovec after 1 year of observation.
BL: baseline;
D30, D90, D180: 30, 90 and 180 days after start of study;
Y1, Y2, Y3: one, two and three years after start of study;
XBL; XD30; XD90; XD180: baseline, 30, 90 and 180 days after start of study for control crossover group;
XY1; XY2: one and two years after start of study for control crossover group.
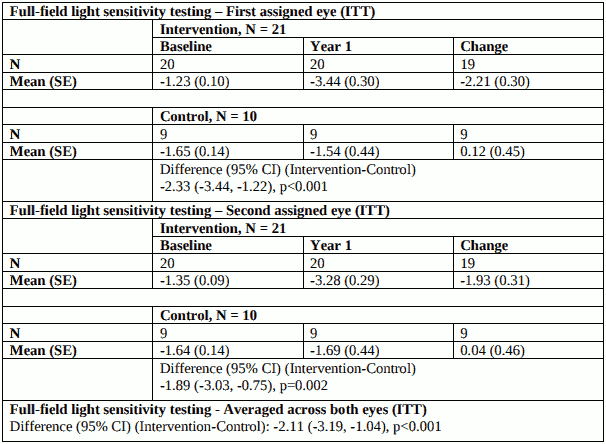
Results of full-field light sensitivity testing at the first study year: white light [Log10(cd.s/m²)] are shown in Table 5 below.
Table 5. Full-field light sensitivity testing:
Improvement in full-field light sensitivity was maintained for up to 3 years after exposure to voretigene neparvovec.
At one year after exposure to voretigene neparvovec, improvement in visual acuity of at least 0.3 LogMAR occurred in 11/20 (55%) of the first-treated eyes and 4/20 (20%) of the second-treated eyes in the intervention group; no one in the control group displayed such an improvement of visual acuity in either the first or second eye.
Voretigene neparvovec is expected to be taken up by cells through heparin sulphate proteoglycan receptors and be degraded by endogenous proteins and DNA catabolic pathways.
Biodistribution of Luxturna was evaluated at three months following subretinal administration in non-human primates. The highest levels of vector DNA sequences were detected in intraocular fluids (anterior chamber fluid and vitreous) of vector-injected eyes. Low levels of vector DNA sequences were detected in the optic nerve of the vector-injected eye, optic chiasm, spleen and liver, and sporadically in the stomach and lymph nodes. In one animal administered with Luxturna at 7.5 × 1011 vg (5 times the recommended per eye dose), vector DNA sequences were detected in colon, duodenum and trachea. Vector DNA sequences were not detected in gonads.
The vector shedding and biodistribution were evaluated in tears from both eyes, serum and whole blood of subjects in the Phase 3 clinical study. In 13/29 (45%) subjects receiving bilateral administrations, Luxturna vector DNA sequences were detected in tear samples; most of these subjects were negative after the day 1 post-injection visit, however, four of these subjects had positive tear samples beyond the first day, one subject up to day 14 post-second eye injection. Vector DNA sequences were detected in serum in 3/29 (10%) subjects, including two with positive tear samples, and only up to day 3 following each injection. Overall, transient and low levels of vector DNA were detected in tear and occasional serum samples from 14/29 (48%) of subjects in the Phase 3 study.
No pharmacokinetic studies with voretigene neparvovec have been conducted in special populations.
Luxturna is injected directly into the eye. Liver and kidney function, cytochrome P450 polymorphisms and ageing are not expected to influence the clinical efficacy or safety of the product. Therefore, no adjustment in dosage is necessary for patients with hepatic or renal impairment.
Ocular histopathology of dog and non-human primate eyes exposed to voretigene neparvovec showed only mild changes, which were mostly related to healing from surgical injury. In an earlier toxicology study, a similar AAV2 vector administered subretinally in dogs at a dose of 10 times the recommended dose resulted in focal retinal toxicity and inflammatory cell infiltrates histologically in regions exposed to the vector. Other findings from voretigene neparvovec non-clinical studies included occasional and isolated inflammatory cells in the retina, with no apparent retinal degeneration. Following a single vector administration, dogs developed antibodies to the AAV2 vector capsid which were absent in naïve non-human primates.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.