LYFNUA Film-coated tablet Ref.[51267] Active ingredients: Gefapixant
Source: European Medicines Agency (EU) Revision Year: 2023 Publisher: Merck Sharp & Dohme B.V., Waarderweg 39, 2031 BN Haarlem, The Netherlands
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Other cough suppressants
ATC code: R05DB29
Mechanism of action
Gefapixant is a selective antagonist of the P2X3 receptor. Gefapixant also has activity against the P2X2/3 receptor subtype. P2X3 receptors are ATP-gated ion channels found on sensory C fibres of the vagus nerve in the airways. C fibres are activated in response to inflammation or chemical irritants. ATP is released from airway mucosal cells under conditions of inflammation. Binding of extracellular ATP to P2X3 receptors is sensed as a damage signal by C fibres. Activation of C fibres, which is sensed by the patient as an urge to cough, initiates a cough reflex. Blockade of ATP signalling through P2X3 receptors reduces excessive sensory-nerve activation and excessive cough induced by extracellular ATP.
Clinical efficacy and safety
The efficacy of Lyfnua for the treatment of refractory or unexplained chronic cough was studied in two 52-week, multicentre, randomised, double-blind, placebo-controlled studies of adults with either refractory or unexplained chronic cough. Refractory chronic cough (RCC) was defined as cough associated with a co-morbid condition (e.g., asthma, gastroesophageal reflux disease, or upper airway cough syndrome) that persisted despite adequate treatment of the co-morbid condition. Unexplained chronic cough (UCC) was defined as cough that was not associated with a co-morbid condition despite a thorough clinical evaluation.
The primary objective of both Phase III studies was to assess Lyfnua efficacy in reducing 24-hour cough frequency relative to placebo. Reduction in awake cough frequency and cough-specific quality of life were secondary objectives. In both studies, patients were randomised to twice daily doses of Lyfnua 45 mg, 15 mg, or placebo. The primary efficacy period for COUGH-1 (NCT03449134) was 12 weeks followed by a blinded extension period of 40 weeks. The primary efficacy period for COUGH-2 (NCT03449147) was 24 weeks, followed by a blinded extension period of 28 weeks.
Patients enrolled in COUGH-1 and COUGH-2 were current non-smokers, not on angiotensin converting enzyme (ACE) inhibitors, diagnosed with RCC or UCC, and had chronic cough for greater than 1 year. Most patients were female (75%), white (80%), and from Europe (53%) with a mean age of 58 years (range 19 to 89) and 7% of patients were older than 75 years. A total of 61.5% of patients were diagnosed with RCC, 38.5% with UCC, and the mean duration of chronic cough was 11 years.
Cough frequency
In COUGH-1 and COUGH-2, patients treated with Lyfnua 45 mg twice daily demonstrated a significant reduction in 24-hour cough frequency compared with placebo (Table 2). The reduction in the 24-hour cough frequency was observed by Week 4 and persisted throughout the primary efficacy period (12 weeks in COUGH-1 and 24 weeks in COUGH-2; Figure 1).
The gefapixant 15 mg twice daily group did not demonstrate a significant reduction in 24-hour cough frequency in either study.
Table 2. 24-hour cough frequency results for Lyfnua 45 mg twice daily (COUGH-1 and COUGH-2):
| COUGH-1 | COUGH-2 | |||
|---|---|---|---|---|
| Lyfnua | Placebo | Lyfnua | Placebo | |
| N | 243 | 243 | 439 | 435 |
| Primary Efficacy Endpoint | ||||
| 24-Hour Cough Frequency (coughs per hour) | ||||
| Baseline (geometric mean) | 18.24 | 22.83 | 18.55 | 19.48 |
| Week 12 (COUGH-1) or Week 24 (COUGH-2) (geometric mean) | 7.05 | 10.33 | 6.83 | 8.34 |
| Week 12 (COUGH-1) or Week 24 (COUGH-2) (%-reduction from baseline) | -61.35 | -54.77 | -63.17 | -57.19 |
Reduction Relative to Placebo
(-reduction and 95 CI)†
| -18.52 (-32.76, -1.28) | -13.29 (-24.74, -0.10) | |||
| p-value | 0.036 | 0.048 |
N = Number of participants included in the analysis. CI = Confidence Interval.
† Missing baseline values were imputed based on gender and region, followed by multiple imputation of the missing data (m = 50 imputed datasets) for all follow-up visits using treatment, gender, region and the other follow-up visits as covariates. Following imputation, an analysis of covariance (ANCOVA) model was conducted at the time point of interest, adjusting for covariates of treatment, baseline, gender, and region.
Figure 1. Analysis of 24-hour cough frequency over time for Lyfnua 45 mg twice daily (COUGH-1 and COUGH-2):
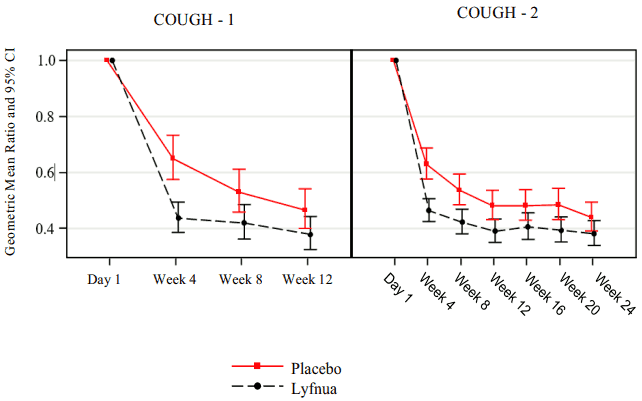
Cough-specific quality of life COUGH-2 was specifically designed to assess the impact of Lyfnua on cough-specific quality of life relative to placebo as measured by the Leicester Cough Questionnaire (LCQ) (possible score ranges from 3 to 21, with higher scores indicating a better quality of life). A ≥1.3 point increase from baseline in the LCQ total score was defined as clinically meaningful. In COUGH-2, the odds of having a clinically meaningful improvement in cough-specific quality of life were significantly greater in the Lyfnua 45 mg treatment group than in the placebo group as measured at Week 24 (see Table 3).
Table 3. Cough-specific quality of life for Lyfnua 45 mg twice daily (COUGH-2): proportion of patients with ≥1.3 point increase from baseline in LCQ total score at Week 24:
| Lyfnua | Placebo | |
|---|---|---|
| N | 439 | 435 |
| Responders* (%) | 75.7 | 68.1 |
| Estimated odds ratio vs. placebo (95% CI)† | 1.46 (1.07, 1.99) | |
| Estimated difference† vs. placebo (95% CI)†† | 7.63 (1.34, 13.76) | |
| p-value† | 0.016 |
N = Number of subjects with available data at Week 24.
* Percent responders at Week 24. Number of responders was calculated by averaging over multiple imputations; there were approximately 332 and 296 responders in Lyfnua and placebo arm, respectively.
CI = Confidence Interval. LCQ = Leicester Cough Questionnaire.
† Missing baseline values were imputed based on gender and region, followed by multiple imputation of the missing data (m = 50 imputed dataset) for all follow-up visits using treatment, gender, region, and the other follow-up visits as covariates. Following imputation, logistic regression was conducted on the dichotomized scores at the time point of interest, adjusting for covariates of treatment, baseline LCQ total (continuous) score, gender, and region.
†† Based on the bootstrap method.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with Lyfnua (gefapixant) in all subsets of the paediatric population in treatment of unexplained or chronic refractory cough (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
The pharmacokinetics of gefapixant were studied in healthy adults and in adults with RCC or UCC and were similar between these two populations. The steady-state mean plasma AUC and peak concentration (Cmax) are 4,144 ng∙hr/mL and 531 ng/mL with gefapixant 45 mg twice daily treatment. Steady state is achieved within 2 days, with an accumulation ratio of 1.4- to 1.5-fold.
Absorption
Following oral administration of gefapixant, the time to achieve peak plasma concentrations (Tmax) ranged from 1 to 4 hours. Exposure increases are dose-proportional following multiple doses up to 300 mg twice daily. The fraction absorbed for gefapixant is at least 78%.
Effect of food
Relative to fasting conditions, oral administration of a single dose of gefapixant 50 mg with a standard high fat and high calorie meal had no effect on the AUC or Cmax of gefapixant. Distribution Based on population pharmacokinetic analyses, the mean steady-state apparent volume of distribution is estimated to be 138 L following oral administration of a 45 mg dose.
In vitro, gefapixant exhibits low plasma protein binding (55%) and has a blood-to-plasma ratio of 1.1. Based on preclinical studies, gefapixant has low CNS penetration.
Biotransformation
Hepatic metabolism is a minor route of gefapixant elimination, involving oxidation and glucuronidation. Following oral administration of [14C] gefapixant, 14% of the administered dose was recovered as metabolites in the urine and faeces. Unchanged gefapixant is the major drug-related component in plasma (87%), and each circulating metabolite accounted for less than 10% of the total radioactivity detected.
Elimination
Renal excretion is the major route of elimination of gefapixant and involves both passive renal filtration and active transport mechanisms. Gefapixant is recovered in urine as parent (~64%) or metabolites (~12%), and the remainder is recovered in feces as parent (~20%) or metabolites (~2%). Active renal secretion is estimated to account for ≤50% of total elimination. In vitro, gefapixant is a substrate of MATE1, MATE2K, P-gp, and BCRP transporters. Gefapixant has a terminal half-life (t½) of 6-10 hours.
Special populations
Renal impairment
Renal excretion is the major route of elimination of gefapixant. Mild or moderate renal impairment (eGFR ≥30 mL/minute/1.73 m²) does not have a clinically meaningful effect on the exposure of gefapixant.
In a population pharmacokinetic analysis including patients with refractory or unexplained chronic cough, the mean AUC and Cmax of gefapixant were predicted to increase by 89% and 54%, respectively, in patients with severe renal impairment (eGFR <30 mL/minute/1.73 m²) compared to those with normal renal function. To maintain similar systemic exposures to those with normal renal function, dose adjustment is recommended (see section 4.2).
Hepatic impairment
Hepatic metabolism is a minor route of elimination. Most of an oral dose was recovered as unchanged parent in the urine (64%) or faeces (20%). A dedicated study in subjects with hepatic impairment was not conducted, because hepatic impairment is not likely to have a clinically meaningful effect on exposure (see section 4.2).
Effects of age, body weight, gender, ethnicity, and race
Based on a population pharmacokinetic analysis, age, body weight, gender, ethnicity, and race do not have a clinically meaningful effect on the pharmacokinetics of gefapixant.
Drug Interactions
Effects of other medicinal products on the pharmacokinetics of gefapixant Hepatic metabolism is a minor pathway for gefapixant elimination, and the potential for clinically meaningful drug interactions for gefapixant with co-administration of inhibitors or inducers of cytochrome P450 (CYP) or uridine 5'-diphosphoglucuronic acid glucuronosyl transferase (UGT) enzymes is low.
Concomitant use of a proton pump inhibitor, omeprazole, did not have a clinically meaningful effect on gefapixant pharmacokinetics.
Based on in vitro studies, gefapixant is a substrate of efflux transporters multidrug and toxin extrusion 1 (MATE1), MATE2K, P-glycoprotein (P-gp), and breast cancer resistance protein (BCRP). In a Phase 1 clinical study, a single dose of the MATE1/MATE2K inhibitor pyrimethamine increased gefapixant AUC by 24%, an amount that is not clinically meaningful, and did not affect gefapixant Cmax.
Effects of gefapixant on the pharmacokinetics of other medicinal products
Based on in vitro studies, the potential of gefapixant to cause CYP inhibition or induction is low, and therefore it is unlikely that gefapixant would affect the CYP-mediated metabolism of other drugs. Gefapixant is an inhibitor of MATE1, MATE2K, and organic anion-transporting polypeptide 1B1 (OATP1B1) and OATP1B3 in vitro. However, the risk of clinically meaningful drug interactions via inhibition of these transporters is low for gefapixant administered at 45 mg twice daily. The clinical relevance of in vitro inhibition of organic cation transporter 1 (OCT1) by gefapixant is not established. In a Phase 1 clinical study, multiple doses of gefapixant 45 mg did not affect exposure of the OATP1B substrate pitavastatin.
5.3. Preclinical safety data
Repeat dose toxicity
Crystalluria occurred in laboratory animals dosed with gefapixant and the majority of the urinary crystals were confirmed to be composed of gefapixant.
In a six month repeat-dose toxicity study in rats, microscopic changes in the kidney (distended tubules due to presence of crystalline material, degeneration of epithelial cells lining tubules and inflammation in the interstitium), ureter (dilatation and inflammation) and bladder (transitional cell hyperplasia) were observed at 9 times the exposure in humans at the maximum recommended human dose (MRHD).
In a nine-month repeat-dose oral toxicity study in dogs, crystals were observed in the urine and microscopic observation of focal, minimal tubular degeneration, involving occasional cortical tubules was observed in one male dog at 35 times the exposure in humans at the MRHD.
Carcinogenicity
Carcinogenicity studies in rats (2-years in duration) and rasH2 transgenic mice (6-months in duration) with gefapixant showed no evidence of carcinogenic potential (no treatment related tumours) at exposures up to 9-times (rats) and 4-times (mice) the exposures at the MRHD.
Mutagenesis
Gefapixant was not genotoxic in a battery of in vitro or in vivo assays including microbial mutagenesis, chromosomal aberration in human peripheral blood lymphocytes and in the in vivo rat micronucleus test.
Reproductive toxicity
In animal reproduction studies, oral administration of gefapixant to pregnant rats and rabbits during the period of organogenesis showed no evidence of teratogenicity or embryo-fetal lethality at exposures (AUC) that were 6-times (rats) and 34-times (rabbits) the exposure at the MRHD. A slight reduction in rat fetal weights, which was associated with maternal toxicity, was observed at an exposure approximately 11-times the exposure at the MRHD.
Studies in pregnant rats and rabbits showed that gefapixant is transferred to the foetus through the placenta, with foetal plasma concentrations of up to 21% (rats) and 25% (rabbits) that of maternal concentrations observed on gestation day 20.
In a lactation study, gefapixant was excreted in milk of lactating rats when administered orally (up to 9-times the exposure at the MRHD) on lactation day 10, with milk concentrations 4 times that of maternal plasma concentrations observed 1-hour post dose on lactation day 10. There were no effects on fertility, mating performance or early embryonic development when gefapixant was administered to female and male rats up to 9-times the exposure at the MRHD.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.