LYNPARZA Film-coated tablet Ref.[8404] Active ingredients: Olaparib
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: AstraZeneca AB, SE-151 85 Södertälje, Sweden
Pharmacodynamic properties
Pharmacotherapeutic group: Other antineoplastic agents
ATC code: L01XK01
Mechanism of action and pharmacodynamic effects
Olaparib is a potent inhibitor of human poly (ADP-ribose) polymerase enzymes (PARP-1, PARP-2, and PARP-3), and has been shown to inhibit the growth of selected tumour cell lines in vitro and tumour growth in vivo either as a standalone treatment or in combination with established chemotherapies or new hormonal agents (NHA).
PARPs are required for the efficient repair of DNA single strand breaks and an important aspect of PARP-induced repair requires that after chromatin modification, PARP auto-modifies itself and dissociates from the DNA to facilitate access for base excision repair (BER) enzymes. When olaparib is bound to the active site of DNA-associated PARP it prevents the dissociation of PARP and traps it on the DNA, thus blocking repair. In replicating cells this also leads to the formation of DNA double-strand breaks (DSBs) when replication forks meet the PARP-DNA adducts. In normal cells, homologous recombination repair (HRR) pathway is effective at repairing these DNA DSBs. In cancer cells lacking critical functional components for efficient HRR such as BRCA1 or 2, DNA DSBs cannot be repaired accurately or effectively, leading to substantial homologous recombination deficiency (HRD). Instead, alternative and error-prone pathways are activated, such as the classical non-homologous end joining (NHEJ) pathway, leading to a high degree of genomic instability. After a number of rounds of replication, genomic instability can reach insupportable levels and result in cancer cell death, as cancer cells already have a high DNA damage load relative to normal cells. HRR pathway may be compromised by other mechanisms, although the causative aberrancy and penetrance are not fully elucidated. Absence of fully functional HRR pathway is one of the key determinants of platinum sensitivity in ovarian and possibly other cancers.
In BRCA1/2-deficient in vivo models, olaparib given after platinum treatment resulted in a delay in tumour progression and an increase in overall survival compared to platinum treatment alone that correlated with the period of olaparib maintenance treatment.
Combined anti-tumour effect with NHAs
Pre-clinical studies in prostate cancer models reported a combined anti-tumour effect when PARP inhibitors and next-generation hormonal agents are administered together. PARP is involved in positive co-regulation of androgen receptor (AR) signalling, which leads to enhanced AR target gene suppression when PARP/AR signalling is co-inhibited. Other pre-clinical studies reported that treatment with NHAs inhibit the transcription of some HRR genes, therefore, inducing HRR deficiency and increased sensitivity to PARP inhibitors via non-genetic mechanisms.
Detection of BRCA1/2 mutations
Genetic testing should be conducted by an experienced laboratory using a validated test. Local or central testing of blood and/or tumour samples for germline and/or somatic BRCA1/2 mutations have been used in different studies. DNA obtained from a tissue or blood sample has been tested in most of the studies, with testing of ctDNA being used for exploratory purposes. Depending on the test used and the international classification consensus, the BRCA1/2 mutations have been classified as deleterious/suspected deleterious or pathogenic/likely pathogenic. Homologous recombination deficiency (HRD) positive status can be defined by detection of a BRCA1/2 mutation classified as deleterious/suspected deleterious or pathogenic/likely pathogenic. Detection of these mutations could be combined with positive HRD score (below) to determine HRD positive status.
Detection of genomic instability
HR deficiency-associated genomic alterations that have been investigated in Paola-1 include genome-wide loss of heterozygosity, telomeric allelic imbalance and large-scale transition, which are continuous measures with pre-defined criteria and score. Composite genomic instability score (GIS, also called HRD score) is determined when the combined measures and respective scores are used to assess the extent of specific genomic aberrations accumulated in tumour cells. Lower score defines lower likelihood of HR deficiency of tumour cells and higher score determines higher likelihood of HR deficiency of tumour cells at the time of the sample collection relative to exposure to DNA damaging agents. Validated cut-offs should be used to determine GIS positive status.
HRD positive status can be defined by a composite GIS score for HR deficiency-associated genomic alterations tested by an experienced laboratory using a validated test.
Clinical efficacy and safety
First-line maintenance treatment of BRCA-mutated advanced ovarian cancer
SOLO1 Study
The safety and efficacy of olaparib as maintenance therapy were studied in patients with newly diagnosed advanced (FIGO Stage III-IV) high-grade serous or endometrioid BRCA1/2 mutated (BRCA1/2m) ovarian cancer following completion of first-line platinum-based chemotherapy in a Phase III randomised, double-blind, placebo-controlled, multicentre trial. In this study 391 patients were randomised 2:1 to receive either Lynparza (300 mg [2 × 150 mg tablets] twice daily) or placebo. Patients were stratified by response to first-line platinum chemotherapy; complete response (CR) or partial response (PR). Treatment was continued until radiological progression of the underlying disease, unacceptable toxicity or for up to 2 years. For patients who remained in complete clinical response (i.e. no radiological evidence of disease), the maximum duration of treatment was 2 years; however, patients who had evidence of disease that remained stable (i.e. no evidence of disease progression) could continue to receive Lynparza beyond 2 years.
Patients with germline or somatic BRCA1/2 mutations were identified prospectively either from germline testing in blood via a local test (n=208) or central test (n=181) or from testing a tumour sample using a local test (n=2). By central germline testing, deleterious or suspected deleterious mutations were identified in 95.3% (365/383) and 4.7% (18/383) of patients, respectively. Large rearrangements in the BRCA1/2 genes were detected in 5.5% (21/383) of the randomised patients. The gBRCAm status of patients enrolled via local testing was confirmed retrospectively by central testing. Retrospective testing of patients with available tumour samples was performed using central testing and generated successful results in 341 patients, of which 95% had an eligible mutation (known [n=47] or likely pathogenic [n=277]) and 2 gBRCAwt patients were confirmed to have sBRCAm only. There were 389 patients who were germline BRCA1/2m and 2 who were somatic BRCA1/2m in SOLO1.
Demographic and baseline characteristics were generally well balanced between the olaparib and placebo treatment arms. Median age was 53 years in both arms. Ovarian cancer was the primary tumour in 85% of the patients. The most common histological type was serous (96%), endometrioid histology was reported in 2% of the patients. Most patients were ECOG performance status 0 (78%), there are no data in patients with performance status 2 to 4. Sixty-three percent (63%) of the patients had upfront debulking surgery and of these the majority (75%) had no macroscopic residual disease. Interval debulking surgery was performed in 35% of the patients and of these 82% had no macroscopic residual disease reported. Seven patients, all stage IV, had no cytoreductive surgery. All patients had received first-line platinum-based therapy. There was no evidence of disease at study entry (CR), defined by the investigator as no radiological evidence of disease and cancer antigen 125 (CA-125) within normal range, in 73% and 77% of patients in the olaparib and placebo arms, respectively. PR, defined as the presence of any measurable or non-measurable lesions at baseline or elevated CA-125, was reported in 27% and 23% of patients in the olaparib and placebo arms, respectively. Ninety three percent (93%) of patients were randomised within 8 weeks of their last dose of platinum-based chemotherapy. Patients who had been treated with bevacizumab were excluded from the study, therefore there are no safety and efficacy data on olaparib patients who had previously received bevacizumab. There are very limited data in patients with a somatic BRCA mutation.
The primary endpoint was progression-free survival (PFS) defined as time from randomisation to progression determined by investigator assessment using modified Response Evaluation Criteria in Solid Tumors (RECIST) 1.1, or death. Secondary efficacy endpoints included time from randomisation to second progression or death (PFS2), overall survival (OS), time from randomisation to discontinuation of treatment or death (TDT), time from randomisation to first subsequent anti-cancer therapy or death (TFST) and health related quality of life (HRQoL). Patients had tumour assessments at baseline and every 12 weeks for 3 years, and then every 24 weeks relative to date of randomisation, until objective radiological disease progression.
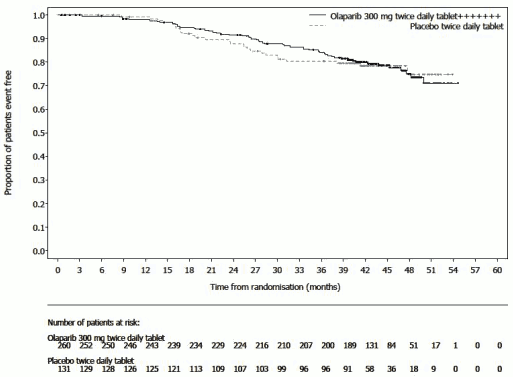
The study demonstrated a clinically relevant and statistically significant improvement in investigator assessed PFS for olaparib compared to placebo. The investigator assessment of PFS was supported with a blinded independent central radiological (BICR) review of PFS. A descriptive analysis performed at seven years after the last patient was randomized demonstrated a clinically meaningful benefit in OS that numerically favoured the olaparib arm. Efficacy results are presented in Table 3 and Figures 1 and 2.
Table 3. Efficacy results for newly diagnosed patients with BRCA1/2m advanced ovarian cancer in SOLO1:
| Olaparib 300 mg bd | Placeboc | |
|---|---|---|
| PFS (51% maturity)a | ||
| Number of events: Total number of patients (%) | 102:260 (39) | 96:131 (73) |
| Median time (months) | NR | 13.8 |
| HR (95% CI)b | 0.30 (0.23-0.41) | |
| P value (2-sided) | p<0.0001 | |
| PFS2 (31% maturity) | ||
| Number of events: Total number of patients (%) | 69:260 (27) | 52:131 (40) |
| Median time (months) | NR | 41.9 |
| HR (95% CI)c | 0.50 (0.35-0.72) | |
| P value (2-sided) | p=0.0002 | |
| OS (38% maturity)d | ||
| Number of events: Total number of patients (%) | 84:260 (32) | 65:131 (50) |
| Median time (months) | NR | 75.2 |
| HR (95% CI)b | 0.55 (0.40-0.76) | |
| TFST (60% maturity) | ||
| Number of events: Total number of patients (%) | 135:260 (52) | 98:131 (75) |
| Median time (months) | 64.0 | 15.1 |
| HR (95% CI)c | 0.37 (0.28-0.48) | |
a Based on Kaplan-Meier estimates, the proportion of patients that were progression free at 24 and 36 months were 74% and 60% for olaparib versus 35% and 27% for placebo; the median follow-up time was 41 months for both the olaparib and placebo arms.
b A value <1 favours olaparib. The analysis was performed using a Cox proportional hazards model including response to previous platinum chemotherapy (CR or PR) as a covariate.
c Of the 97 patients on the placebo arm who received subsequent therapy, 58 (60%) received a PARP inhibitor.
d Based on Kaplan-Meier estimates, the proportion of patients that were alive 84 months was 67% for olaparib versus 47% for placebo.
bd Twice daily; NR Not reached; CI Confidence interval; PFS Progression-free survival; PFS2 Time to second progression or death; OS Overall survival; TFST Time from randomisation to first subsequent anti-cancer therapy or death.
Figure 1. SOLO1: Kaplan-Meier plot of PFS in newly diagnosed patients with BRCA1/2m advanced ovarian cancer (51% maturity – investigator assessment):
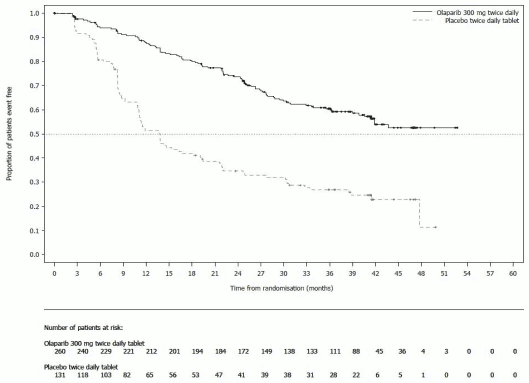
Figure 2. SOLO1: Kaplan-Meier plot of OS in newly diagnosed patients with BRCA1/2m advanced ovarian cancer (38% maturity):
Consistent results were observed in the subgroups of patients by evidence of the disease at study entry. Patients with CR defined by the investigator had HR 0.34 (95% CI 0.24-0.47); median PFS not reached on olaparib vs 15.3 months on placebo. At 24 and 36 months, respectively, 68% and 45% patients remained in CR in the olaparib arm, and 34% and 22% of patients in the placebo arm. Patients with PR at study entry had PFS HR 0.31 (95% CI 0.18, 0.52; median PFS 30.9 months on olaparib vs 8.4 months on placebo). Patients with PR at study entry either achieved CR (15% in olaparib arm and 4% in the placebo arm at 24 months, remained in CR at 36 months) or had further PR/stable disease (43% in olaparib arm and 15% in the placebo arm at 24 months; 17% in olaparib arm and 15% in placebo arm at 36 months). The proportion of patients who progressed within 6 months of the last dose of platinum-based chemotherapy was 3.5% for olaparib and 8.4% for placebo.
Maintenance treatment of platinum-sensitive relapsed (PSR) ovarian cancer
SOLO2 Study
The safety and efficacy of olaparib as maintenance therapy were studied in a Phase III randomised, double-blind, placebo-controlled trial in patients with germline BRCA1/2-mutated PSR ovarian, fallopian tube or primary peritoneal cancer. The study compared the efficacy of Lynparza maintenance treatment (300 mg [2 × 150 mg tablets] twice daily) taken until progression with placebo treatment in 295 patients with high-grade serous or endometrioid PSR ovarian cancer (2:1 randomisation: 196 olaparib and 99 placebo) who were in response (CR or PR) following completion of platinum-containing chemotherapy.
Patients who have received two or more platinum-containing regimens and whose disease had recurred >6 months after completion of penultimate platinum-based chemotherapy were enrolled. Patients could not have received prior olaparib or other PARP inhibitor treatment. Patients could have received prior bevacizumab, except in the regimen immediately prior to randomisation.
All patients had evidence of gBRCA1/2m at baseline. Patients with BRCA1/2 mutations were identified either from germline testing in blood via a local test or by central testing at Myriad or from testing a tumour sample using a local test. Large rearrangements in the BRCA1/2 genes were detected in 4.7% (14/295) of the randomised patients.
Demographic and baseline characteristics were generally well balanced between the olaparib and placebo arms. Median age was 56 years in both arms. Ovarian cancer was the primary tumour in >80% of the patients. The most common histological type was serous (>90%), endometrioid histology was reported in 6% of the patients. In the olaparib arm 55% of the patients had only 2 prior lines of treatment with 45% receiving 3 or more prior lines of treatment. In the placebo arm 61% of patients had received only 2 prior lines with 39% receiving 3 or more prior lines of treatment. Most patients were ECOG performance status 0 (81%), there are no data in patients with performance status 2 to 4. Platinum-free interval was >12 months in 60% and >6-12 months in 40% of the patients. Response to prior platinum chemotherapy was complete in 47% and partial in 53% of the patients. In the olaparib and placebo arms, 17% and 20% of patients had prior bevacizumab, respectively.
The primary endpoint was PFS determined by investigator assessment using RECIST 1.1. Secondary efficacy endpoints included PFS2; OS, TDT, TFST, TSST; and HRQoL.
The study met its primary objective demonstrating a statistically significant improvement in investigator assessed PFS for olaparib compared with placebo with a HR of 0.30 (95% CI 0.22-0.41; p<0.0001; median 19.1 months olaparib vs 5.5 months placebo). The investigator assessment of PFS was supported with a blinded independent central radiological review of PFS (HR 0.25; 95% CI 0.18-0.35; p<0.0001; median 30.2 months for olaparib and 5.5 months placebo). At 2 years, 43% olaparib-treated patients remained progression free compared with only 15% placebo-treated patients.
A summary of the primary objective outcome for patients with gBRCA1/2m PSR ovarian cancer in SOLO2 is presented in Table 4 and Figure 3.
Table 4. Summary of primary objective outcome for patients with gBRCA1/2m PSR ovarian cancer in SOLO2:
| Olaparib 300 mg tablet bd | Placebo | |
|---|---|---|
| PFS (63% maturity) | ||
| Number of events: Total number of patients (%) | 107:196 (55) | 80:99 (81) |
| Median time (months) (95% CI) | 19.1 (16.3-25.7) | 5.5 (5.2-5.8) |
| HR (95% CI)a | 0.30 (0.22-0.41) | |
| P value (2-sided) | p<0.0001 | |
a HR = Hazard Ratio. A value <1 favours olaparib. The analysis was performed using a Cox proportional hazard model including response to previous platinum chemotherapy (CR or PR), and time to disease progression (>6-12 months and >12 months) in the penultimate platinum-based chemotherapy as covariates.
bd Twice daily; PFS progression-free survival; CI confidence interval
Figure 3. SOLO2: Kaplan-Meier plot of PFS in patients with gBRCA1/2m PSR ovarian cancer (63% maturity – investigator assessment):
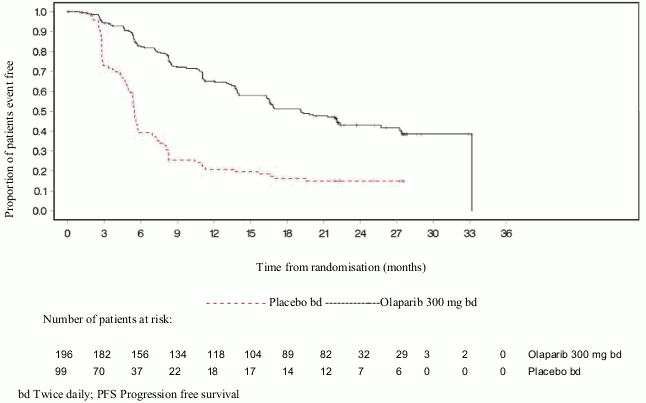
bd Twice daily; PFS Progression free survival
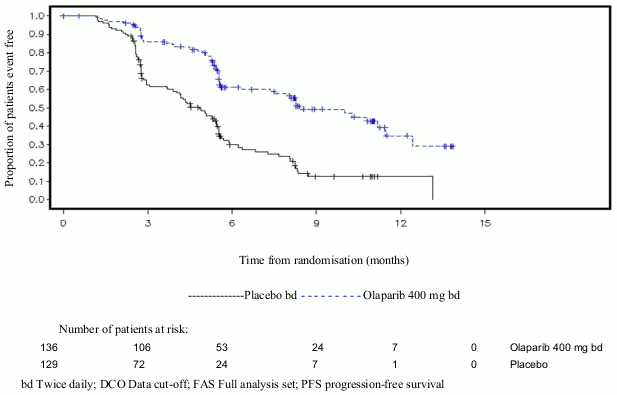
At the final analysis of OS (61% maturity) the HR was 0.74 (95% CI 0.54-1.00; p=0.0537; median 51.7 months for olaparib vs 38.8 months for placebo) which did not reach statistical significance. The secondary endpoints TFST and PFS2 demonstrated a persistent and statistically significant improvement for olaparib compared with placebo. Results for OS, TFST and PFS2 are presented in Table 5 and Figure 4.
Table 5. Summary of key secondary objective outcomes for patients with gBRCA1/2m PSR ovarian cancer in SOLO2:
| Olaparib 300 mg tablet bd | Placebo | |
|---|---|---|
| OS (61% maturity) | ||
| Number of events: Total number of patients (%) | 116:196 (59) | 65:99 (66) |
| Median time (95% CI), months | 51.7 (41.5, 59.1) | 38.8 (31.4, 48.6) |
| HR (95% CI)a | 0.74 (0.54-1.00) | |
| P value (2-sided) | p=0.0537 | |
| TFST (71% maturity) | ||
| Number of events: Total number of patients (%) | 139:196 (71) | 86:99 (87) |
| Median time (months) (95% CI) | 27.4 (22.6-31.1) | 7.2 (6.3-8.5) |
| HR (95% CI)a | 0.37 (0.28-0.48) | |
| P value* (2-sided) | p<0.0001 | |
| PFS2 (40% maturity) | ||
| Number of events: Total number of patients (%) | 70:196 (36) | 49:99 (50) |
| Median time (months) (95% CI) | NR (24.1-NR) | 18.4 (15.4-22.8) |
| HR (95% CI)a | 0.50 (0.34-0.72) | |
| P value (2-sided) | p=0.0002 | |
* Not controlled for multiplicity.
a HR = Hazard Ratio. A value <1 favours olaparib. The analysis was performed using a Cox proportional hazard model including response to previous platinum chemotherapy (CR or PR), and time to disease progression (>6-12 months and >12 months) in the penultimate platinum-based chemotherapy as covariates.
bd Twice daily; NR not reached; CI confidence interval; PFS2 time from randomisation to second progression or death; TFST Time from randomisation to start of first subsequent therapy or death.
Figure 4. SOLO2: Kaplan-Meier plot of OS in patients with gBRCA1/2m PSR ovarian cancer (61% maturity):
mong the patients entering the trial with measurable disease (target lesions at baseline), an objective response rate of 41% was achieved in the Lynparza arm versus 17% on placebo. Of patients treated with Lynparza, who entered the study with evidence of disease (target or non-target lesions at baseline), 15.0% experienced complete response compared with 9.1% of patients on placebo.
At the time of the analysis of PFS the median duration of treatment was 19.4 months for olaparib and 5.6 months for placebo. The majority of patients remained on the 300 mg bd starting dose of olaparib. The incidence of dose interruptions, reductions, discontinuations due to an adverse event was 45.1%, 25.1% and 10.8%, respectively. Dose interruptions occurred most frequently in the first 3 months and dose reductions in the first 3-6 months of treatment. The most frequent adverse reactions leading to dose interruption or dose reduction were anaemia, nausea and vomiting.
Patient-reported outcome (PRO) data indicate no difference for the olaparib-treated patients as compared to placebo as assessed by the change from baseline in the TOI of the FACT-O.
Study 19 (D0810C00019)
The safety and efficacy of olaparib as a maintenance therapy in the treatment of PSR ovarian, including fallopian tube or primary peritoneal cancer patients, following treatment with two or more platinum-containing regimens, were studied in a large Phase II randomised, double-blind, placebo-controlled trial (Study 19). The study compared the efficacy of Lynparza maintenance treatment taken until progression with placebo treatment in 265 (136 olaparib and 129 placebo) PSR high grade serous ovarian cancer patients who were in response (CR or PR) following completion of platinum-containing chemotherapy. The primary endpoint was PFS based on investigator assessment using RECIST 1.0. Secondary efficacy endpoints included OS, disease control rate (DCR) defined as confirmed CR/PR + SD (stable disease), HRQoL and disease related symptoms. Exploratory analyses of TFST and TSST were also performed.
Patients whose disease had recurred >6 months after completion of penultimate platinum-based chemotherapy were enrolled. Enrolment did not require evidence of BRCA1/2 mutation (BRCA mutation status for some patients was determined retrospectively). Patients could not have received prior olaparib or other PARP inhibitor treatment. Patients could have received prior bevacizumab, except in the regimen immediately prior to randomisation. Retreatment with olaparib was not permitted following progression on olaparib.
Patients with BRCA1/2 mutations were identified either from germline testing in blood via a local test or by central testing at Myriad or from testing a tumour sample using a test performed by Foundation Medicine. Large rearrangements in the BRCA1/2 genes were detected in 7.4% (10/136) of the randomised patients.
Demographic and baseline characteristics were generally well balanced between the olaparib and placebo arms. Median age was 59 years in both arms. Ovarian cancer was the primary tumour in 86% of the patients. In the olaparib arm 44% of the patients had only 2 prior lines of treatment with 56% receiving 3 or more prior lines of treatment. In the placebo arm 49% of patients had received only 2 prior lines with 51% receiving 3 or more prior lines of treatment. Most patients were ECOG performance status 0 (77%), there are no data in patients with performance status 2 to 4. Platinum-free interval was >12 months in 60% and 6-12 months in 40% of the patients. Response to prior platinum chemotherapy was complete in 45% and partial in 55% of the patients. In the olaparib and placebo arms, 6% and 5% of patients had prior bevacizumab, respectively.
The study met its primary objective demonstrating a statistically significant improvement in PFS for olaparib compared with placebo in the overall population with a HR of 0.35 (95% CI 0.25-0.49; p<0.00001; median 8.4 months olaparib vs 4.8 months placebo). At the final OS analysis (data cut off [DCO] 9 May 2016) at 79% maturity, the hazard ratio comparing olaparib with placebo was 0.73 (95% CI 0.55-0.95; p=0.02138 [did not meet pre-specified significance level of <0.0095]; median 29.8 months olaparib versus 27.8 months placebo). In the olaparib-treated group, 23.5% (n=32/136) of patients remained on treatment for ≥2 years as compared with 3.9% (n=5/128) of the patients on placebo. Although patient numbers were limited, 13.2% (n=18/136) of the patients in the olaparib-treated group remained on treatment for ≥5 years as compared with 0.8% (n=1/128) in the placebo group.
Preplanned subgroup analysis identified patients with BRCA1/2-mutated ovarian cancer (n=136, 51.3%; including 20 patients identified with a somatic tumour BRCA1/2 mutation) as the subgroup that derived the greatest clinical benefit from olaparib maintenance monotherapy. A benefit was also observed in patients with BRCA1/2 wild-type/variants of uncertain significance (BRCA1/2 wt/VUS), although of a lesser magnitude. There was no strategy for multiple testing in place for the sub-group analyses.
A summary of the primary objective outcome for patients with BRCA1/2-mutated and BRCA1/2 wt/VUS PSR ovarian cancer in Study 19 is presented in Table 6 and for all patients in Study 19 in Table 6 and Figure 5.
Table 6. Summary of primary objective outcome for all patients and patients with BRCA1/2-mutated and BRCA1/2 wt/VUS PSR ovarian cancer in Study 19:
| All patientsa | BRCA1/2-mutated | BRCA1/2 wt/VUS | ||||
|---|---|---|---|---|---|---|
| Olaparib | Placebo | Olaparib | Placebo | Olaparib | Placebo | |
| PFS – DCO 30 June 2010 | ||||||
| Number of events: Total number of patients (%) | 60:136 (44) | 94:129 (73) | 26:74 (35) | 46:62 (74) | 32:57 (56) | 44:61 (72) |
| Median time (months) (95% CI) | 8.4 (7.4-11.5) | 4.8 (4.0-5.5) | 11.2 (8.3-NR) | 4.3 (3.0-5.4) | 7.4 (5.5-10.3) | 5.5 (3.7-5.6) |
| HR (95% CI)b | 0.35 (0.25-0.49) | 0.18 (0.10–0.31) | 0.54 (0.34-0.85) | |||
| P value (2-sided) | p<0.00001 | p<0.00001 | p=0.00745 | |||
a All patients comprises of the following subgroups: BRCA1/2-mutated, BRCA1/2 wt/VUS and BRCA1/2 status unknown (11 patients with status unknown, not shown as a separate subgroup in table).
b HR = Hazard Ratio. A value <1 favours olaparib. The analysis was performed using a Cox proportional hazards model with factors for treatment, ethnic descent, platinum sensitivity and response to final platinum therapy.
PFS progression-free survival; DCO data cut off; CI confidence interval; NR not reached.
Figure 5. Study 19: Kaplan-Meier plot of PFS in the FAS (58% maturity – investigator assessment) DCO 30 June 2010:
DCO Data cut-off; FAS Full analysis set; PFS progression-free survival
A summary of key secondary objective outcomes for patients with BRCA1/2-mutated and BRCA1/2 wt/VUS PSR ovarian cancer in Study 19 is presented in Table 7 and for all patients in Study 19 in Table 7 and Figure 6.
Table 7. Summary of key secondary objective outcomes for all patients and patients with BRCA1/2-mutated and BRCA1/2 wt/VUS PSR ovarian cancer in Study 19:
| All patientsa | BRCA1/2-mutated | BRCA1/2 wt/VUS | ||||
|---|---|---|---|---|---|---|
| Olaparib | Placebo | Olaparib | Placebo | Olaparib | Placebo | |
| OS – DCO 09 May 2016 | ||||||
| Number of events: Total number of patients (%) | 98:136 (72) | 112:129 (87) | 49:74 (66) | 50:62 (81)c | 45:57 (79) | 57:61 (93) |
| Median time (months) (95% CI) | 29.8 (26.9-35.7) | 27.8 (24.9-33.7) | 34.9 (29.2-54.6) | 30.2 (23.1-40.7) | 24.5 (19.8-35.0) | 26.6 (23.1-32.5) |
| HR (95% CI)b | 0.73 (0.55–0.95) | 0.62 (0.42–0.93) | 0.84 (0.57-1.25) | |||
| P value* (2-sided) | p=0.02138 | p=0.02140 | p=0.39749 | |||
| TFST – DCO 09 May 2016 | ||||||
| Number of events: Total number of patients (%) | 106:136 (78) | 124:128 (97) | 55:74 (74) | 59:62 (95) | 47:57 (83) | 60:61 (98) |
| Median time (months) (95% CI) | 13.3 (11.3-15.7) | 6.7 (5.7-8.2) | 15.6 (11.9-28.2) | 6.2 (5.3-9.2) | 12.9 (7.8-15.3) | 6.9 (5.7-9.3) |
| HR (95% CI)b | 0.39 (0.30–0.52) | 0.33 (0.22-0.49) | 0.45 (0.30-0.66) | |||
| P value* (2-sided) | p<0.00001 | p<0.00001 | p=0.00006 | |||
* There was no strategy for multiple testing in place for the sub-group analyses or for the all patients TFST.
a All patients comprises of the following subgroups: BRCA1/2-mutated, BRCA1/2 wt/VUS and BRCA1/2 status unknown (11 patients with status unknown, not shown as a separate subgroup in table).
b HR = Hazard Ratio. A value <1 favours olaparib. The analysis was performed using a Cox proportional hazards model with factors for treatment, ethnic descent, platinum sensitivity and response to final platinum therapy.
c Approximately a quarter of placebo-treated patients in the BRCA-mutated subgroup (14/62; 22.6%) received a subsequent PARP inhibitor.
OS Overall survival; DCO data cut off; CI confidence interval; TFST time from randomisation to start of first subsequent therapy or death.
Figure 6. Study 19: Kaplan Meier plot of OS in the FAS (79% maturity) DCO 09 May 2016:
DCO Data cut off; FAS Full analysis set; OS Overall survival
At the time of the analysis of PFS the median duration of treatment was 8 months for olaparib and 4 months for placebo. The majority of patients remained on the starting dose of olaparib. The incidence of dose interruptions, reductions and discontinuations due to an adverse event was 34.6%, 25.7% and 5.9%, respectively. Dose interruptions and reductions occurred most frequently in the first 3 months of treatment. The most frequent adverse reactions leading to dose interruption or dose reduction were nausea, anaemia, vomiting, neutropenia and fatigue. The incidence of anaemia adverse reactions was 22.8% (CTCAE grade ≥3 7.4%).
Patient-reported outcome (PRO) data indicate no difference for the olaparib-treated patients as compared to placebo as measured by improvement and worsening rates in the TOI and FACT-O total.
PINION Study
OPINION, a Phase IIIb single arm, multicentre study, investigated olaparib as a maintenance treatment in patients with PSR ovarian, fallopian tube or primary peritoneal cancer following 2 or more lines of platinum based chemotherapy and who did not have a known deleterious or suspected deleterious gBRCA mutation. Patients whose disease was in response (CR or PR) following completion of platinum-based chemotherapy were enrolled. A total of 279 patients were enrolled and received olaparib treatment in this study until disease progression or unacceptable toxicity. Based on central testing 90.7% were confirmed with a non gBRCAm status, in addition 9.7% were identified as sBRCAm.
The primary endpoint was investigator-assessed PFS according to modified RECIST v1.1. Secondary endpoints included OS.
Olaparib, when used as maintenance therapy, demonstrated clinical activity in patients with non-gBRCAm PSR ovarian cancer. At the final overall survival analysis (DCO 17 September 2021), the OS data were 52.3% mature.
A summary of the primary PFS and OS secondary objective outcome for patients with non-gBRCAm PSR ovarian cancer in OPINION is presented in Table 8.
Table 8. Summary of key objective outcome for non-gBRCAm patients with PSR ovarian cancer in OPINION:
| Olaparib tablets 300 mg bd | |
|---|---|
| PFS (75% maturity) (DCO 2 October 2020) | |
| Number of events: total number of patients (%) | 210: 279 (75.3) |
| Median PFS (95% CI), monthsa | 9.2 (7.6, 10.9) |
| OS (52.3% maturity) (DCO 17 September 2021) | |
| Number of events: total number of patients (%) | 146: 279 (52.3) |
| Median OS (95% CI), monthsa | 32.7 (29.5, 35.3) |
a Calculated using the Kaplan-Meier technique.
Confidence intervals for median PFS and OS were derived based on Brookmeyer Crowley method.
bd Twice daily; PFS Progression-free survival; OS Overall survival; DCO Data cut off; CI Confidence interval.
First-line maintenance treatment of HRD positive advanced ovarian cancer
PAOLA-1 Study
PAOLA-1 was a Phase III randomised, double-blind, placebo-controlled, multicentre trial that compared the efficacy and safety of Lynparza (300 mg [2 × 150 mg tablets] twice daily) in combination with bevacizumab (15 mg/kg of body weight given once every 3 weeks as an intravenous infusion) versus placebo plus bevacizumab for the maintenance treatment of advanced (FIGO Stage III-IV) high-grade epithelial ovarian, fallopian tube or primary peritoneal cancer following first-line platinum-based chemotherapy and bevacizumab. Treatment with bevacizumab was for a total of up to 15 months/22 cycles, including the period given with chemotherapy and given as maintenance.
The study randomised 806 patients (2:1 randomisation: 537 olaparib/bevacizumab: 269 placebo/bevacizumab) who had no evidence of disease (NED) due to complete surgical resection, or who were in complete response (CR), or partial response (PR) following completion of first-line platinum-containing chemotherapy and bevacizumab. Patients had completed a minimum of 4 and a maximum of 9 cycles, with the majority (63%) having received 6 cycles of first line platinum-taxane based chemotherapy, including a minimum of 2 cycles of bevacizumab in combination with the 3 last cycles of chemotherapy. The median number of bevacizumab cycles prior to randomisation was 5.
Patients were stratified by first-line treatment outcome (timing and outcome of cytoreductive surgery and response to platinum-based chemotherapy) and tBRCAm status, determined by prospective local testing. Patients continued bevacizumab in the maintenance setting and started treatment with Lynparza after a minimum of 3 weeks and up to a maximum of 9 weeks following completion of their last dose of chemotherapy. Treatment with Lynparza was continued until progression of the underlying disease, unacceptable toxicity or for up to 2 years. Patients who in the opinion of the treating physician could derive further benefit from continuous treatment could be treated beyond 2 years.
Demographic and baseline characteristics were balanced between both arms in the intent to treat (ITT) population and in the biomarker-defined sub-groups by tBRCAm (prospectively and retrospectively defined), GIS and HRD status (defined in this study by a combination of both biomarkers). The median age of patients was 61 years overall. Most patients in both arms were ECOG performance status 0 (70%). Ovarian cancer was the primary tumour in 86% of the patients. The most common histological type was serous (96%) and endometrioid histology was reported in 2% of the patients. Most patients were diagnosed in FIGO stage IIIC (63%). All patients had received first-line platinum- based therapy and bevacizumab. Patients were not restricted by the surgical outcome with 63% having complete cytoreduction at initial or interval debulking surgery and 37% having residual macroscopic disease. Thirty percent (30%) of patients in both arms were tBRCAm at screening. Demographic and baseline characteristics in the biomarker sub-groups were consistent with those in the ITT population. In the HRD-positive subgroup, 65% of patients had complete cytoreduction and 35% of patients had residual macroscopic disease. In the overall patient population enrolled, 30% of patients in both arms were tBRCAm (deleterious/pathogenic mutation) at screening by local testing and for 4% of patients the BRCAm status was unknown. Retrospective analysis of available clinical samples was conducted in 97% of patients to confirm tBRCAm status and investigate genomic instability score as described above. Among non-tBRCAm patients, 29% (19% of the overall population) had positive GIS pre- defined in this study as composite score ≥42. When tBRCAm status and positive GIS were combined, patients with HRD-positive, HRD-negative and HRD unknown status in their tumours represented 48%, 34% and 18% of the overall patient population.
The primary endpoint was progression-free survival (PFS), defined as time from randomisation to progression determined by investigator assessment using modified Response Evaluation Criteria in Solid Tumors (RECIST) 1.1, or death. Secondary efficacy endpoints included time from randomisation to second progression or death (PFS2), overall survival (OS), time from randomisation to first subsequent anti-cancer therapy or death (TFST) and health related quality of life (HRQoL). Patients had RECIST 1.1 tumour assessments at baseline and every 24 weeks (CT/MRI at 12 weeks if clinical or CA 125 progression) for up to 42 months or until objective radiological disease progression.
The study met its primary endpoint in the ITT population demonstrating a statistically significant improvement in investigator assessed PFS for olaparib/bevacizumab compared to placebo/bevacizumab (HR 0.59, 95% CI 0.49-0.72, p<0.0001 with a median of 22.1 months for olaparib/bevacizumab vs 16.6 months for placebo/bevacizumab). This was consistent with a BICR analysis of PFS. However, patients defined as biomarker-positive (tBRCAm, GIS, HRD status positive defined as tBRCAm and/or GIS positive) derived most of the benefit. Final analysis of PFS2 (DCO 22 March 2020, 53% maturity) in the overall population was statistically significant (HR 0.78, 95% CI 0.64-0.95, p=0.0125 with a median of 36.5 months for olaparib/bevacizumab vs 32.6 months for placebo/bevacizumab).
At the final analysis of OS (DCO 22 March 2022) in the HRD status positive patients (tBRCAm and/or GIS), there was a numerical improvement in OS with olaparib/bevacizumab arm vs placebo/bevacizumab arm (see Table 9).
In the tBRCAm as randomised subgroup (241/806 patients) median PFS for the olaparib/bevacizumab arm was 37.2 months vs 22.0 months for the placebo/bevacizumab arm (HR=0.34, 95% CI 0.23, 0.51). At the final overall survival analysis (DCO 22 March 2022), the tBRCAm as randomised subgroup demonstrates a numerical reduction in the risk of death for olaparib/bevacizumab compared to placebo/bevacizumab (HR 0.63; 95% CI 0.41, 0.97).
Efficacy results in other biomarkers subgroup analyses based on retrospectively analysed tumour samples are presented in Table 9.
Table 9. Summary of key efficacy findings for patients with homologous recombination deficiency (HRD) positive status defined by either tBRCAm and/or GIS in advanced ovarian cancer patients in PAOLA-1:
| tBRCAm*c (n=235) | GIS positive (HRD positive excluding tBRCAm)*d (n=152) | HRD positive* (n=387) | ||||
|---|---|---|---|---|---|---|
| Olaparib/ bevacizumab | Placebo/ bevacizumab | Olaparib/ bevacizumab | Placebo/ bevacizumab | Olaparib/ bevacizumab | Placebo/ bevacizumab | |
| PFS, investigator assessment (46% maturity) DCO 22 March 2019a | ||||||
| Number of events: Total number of patients (%) | 44:158 (28) | 52:77 (68) | 43:97 (44) | 40:55 (73) | 87:255 (34) | 92:132 (70) |
| Median time (months) | 37.2 | 18.8 | 28.1 | 16.6 | 37.2 | 17.7 |
| HR (95%) CIb | 0.28 (0.19, 0.42) | 0.43 (0.28, 0.66) | 0.33 (0.25, 0.45) | |||
| PFS2, investigator assessment (40% maturity) DCO 22 March 2020 | ||||||
| Number of events: Total number of patients (%) | 44:158 (28) | 37:77 (48) | 41:97 (42) | 33:55 (60) | 85:255 (33) | 70:132 (53) |
| Median time (months) | NR | 42.2 | 50.3 | 30.1 | 50.3 | 35.4 |
| HR (95%) CIb | 0.53 (0.34, 0.82) | 0.60 (0.38, 0.96) | 0.56 (0.41, 0.77) | |||
| Final OS (42% maturity) DCO 22 March 2022 | ||||||
| Number of events: Total number of patients (%) | 49:158 (31.0) | 37:77 (48.1) | 44:97 (45.4) | 32:55 (58.2) | 93:255 (36.5) | 69:132 (52.3) |
| Median time (months) | 75.2 | 66.9 | NR | 52.0 | 75.2 | 57.3 |
| HR (95%) CIb | 0.57 (0.37, 0.88) | 0.71 (0.45, 1.13) | 0.62 (0.45, 0.85) | |||
* Pre-planned subgroup
a Based on Kaplan-Meier estimates, the proportion of patients that were progression free at 12 and 24 months were 89% and 66% for olaparib/bevacizumab versus 71% and 29% for placebo/bevacizumab.
b A value <1 favours olaparib. The analysis was performed using a Cox proportional hazards model stratified by first line treatment outcome at screening and screening laboratory tBRCA status.
c tBRCAm status by Myriad
d HRD positive excluding tBRCAm was defined as Genomic instability score (GIS) by Myriad ≥42 (pre-specified cut-off)
CI Confidence interval; HR Hazard ratio; NR not reached
Figure 7. PAOLA-1: Kaplan-Meier plot of PFS for patients with advanced ovarian cancer defined as HRD positive in PAOLA-1 (46% maturity – investigator assessment):
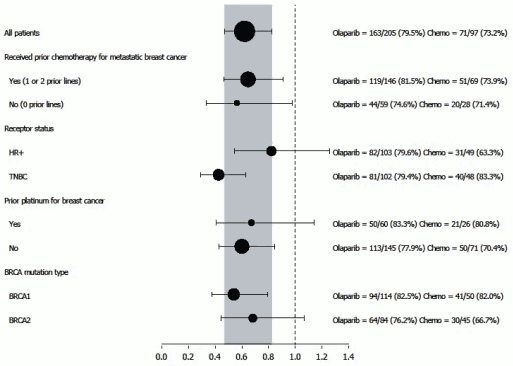
Figure 8. PAOLA-1: Kaplan-Meier Plot, Final Overall Survival by HRD Status Positive (including tBRCAm) (DCO 22 March 2022):
Adjuvant treatment of germline BRCA-mutated high risk early breast cancer
OlympiA
The safety and efficacy of olaparib as adjuvant treatment in patients with germline BRCA1/2 mutations and HER2-negative high risk early breast cancer who had completed definitive local treatment and neoadjuvant or adjuvant chemotherapy was studied in a Phase III randomised, double-blind, parallel group, placebo-controlled, multicentre study (OlympiA). Patients were required to have completed at least 6 cycles of neoadjuvant or adjuvant chemotherapy containing anthracyclines, taxanes or both. Prior platinum for previous cancer (e.g. ovarian) or as adjuvant or neoadjuvant treatment for breast cancer was allowed. High risk early breast cancer patients were defined as follows:
- patients who received prior neoadjuvant chemotherapy: patients with either triple negative breast cancer (TNBC) or hormone receptor positive breast cancer must have had residual invasive cancer in the breast and/or the resected lymph nodes (non-pathologic complete response) at the time of surgery. Additionally, patients with hormone receptor positive breast cancer must have had a CPS&EG score of ≥3 based on pre-treatment clinical and post-treatment pathologic stage (CPS), estrogen receptor (ER) status and histologic grade as shown in Table 10.
Table 10. Early Breast Cancer Stage, Receptor Status and Grade Scoring Requirements for Study Enrolment*:
| Stage/feature | Points | |
|---|---|---|
| Clinical Stage (pre-treatment) | I/IIA | 0 |
| IIB/IIIA | 1 | |
| IIIB/IIIC | 2 | |
| Pathologic Stage (post-treatment) | 0/I | 0 |
| IIA/IIB/IIIA/IIIB | 1 | |
| IIIC | 2 | |
| Receptor status | ER positive | 0 |
| ER negative | 1 | |
| Nuclear grade | Nuclear grade 1-2 | 0 |
| Nuclear grade 3 | 1 | |
* Total score of ≥3 required for patients with hormone receptor positive breast cancer.
- patients who have received prior adjuvant chemotherapy: triple negative breast cancer (TNBC) patients must have had node positive disease or node negative disease with a ≥2 cm primary tumour; HR positive, HER2-negative patients must have had ≥4 pathologically confirmed positive lymph nodes.
Patients were randomised (1:1) to either olaparib 300 mg (2 × 150 mg tablets) twice daily (n=921) or placebo (n=915). Randomisation was stratified by hormone receptor status (HR positive/HER2 negative versus TNBC), by prior neoadjuvant versus adjuvant chemotherapy, and by prior platinum use for current breast cancer (yes versus no). Treatment was continued for up to 1 year, or until disease recurrence, or unacceptable toxicity. Patients with HR positive tumours also received endocrine therapy.
The primary endpoint was invasive disease free survival (IDFS), defined as the time from randomisation to date of first recurrence, where recurrence is defined as invasive loco-regional, distant recurrence, contralateral invasive breast cancer, new cancer or death from any cause. Secondary objectives included OS, distant disease free survival (DDFS, defined as the time from randomisation until evidence of first distant recurrence of breast cancer), the incidence of new primary contralateral breast cancers (invasive and non-invasive), new primary ovarian cancer, new primary fallopian tube cancer and new primary peritoneal cancer, and patient reported outcomes (PRO) using the FACIT- Fatigue and EORTC QLQ-C30 questionnaires.
Central testing at Myriad or local gBRCA testing, if available, was used to establish study eligibility. Patients enrolled based on local gBRCA test results provided a sample for retrospective confirmatory testing. Out of 1836 patients enrolled into OlympiA, 1623 were confirmed as gBRCAm by central testing, either prospectively or retrospectively.
Demographic and baseline characteristics were well balanced between the two treatment arms. The median age was 42 years. Sixty-seven percent (67%) of patients were White, 29% Asian and 2.6% Black. Two patients (0.2%) in the olaparib arm and four patients (0.4%) in the placebo arm were male. Sixty-one percent (61%) of patients were pre-menopausal. Eighty-nine percent (89%) of patients were ECOG performance status 0 and 11% ECOG PS 1. Eighty-two percent (82%) of patients had TNBC and 18% had HR positive disease. Fifty percent (50%) of patients had received prior neoadjuvant and 50% received prior adjuvant chemotherapy. Ninety-four percent (94%) of patients received anthracycline and taxane. Twenty-six percent (26%) of patients overall had received prior platinum for breast cancer. In the olaparib and placebo arms, 87% and 92% of patients with HR positive disease were receiving concomitant endocrine therapy, respectively. Overall, 89.5% of patients with HR positive disease received an endocrine therapy, which included letrozole (23.7%), tamoxifen (40.9%), anastrozole (17.2%), or exemestane (14.8%).
The study met its primary endpoint demonstrating a statistically significant improvement in IDFS in the olaparib arm compared with the placebo arm. Two hundred and eighty-four (284) patients had IDFS events, this represented 12% of patients in the olaparib arm (distant 8%, local/regional 1.4%, contralateral invasive breast cancer 0.9%, non-breast second primary malignancies 1.2%, death 0.2%) and 20% of patients in the placebo arm (distant 13%, local/regional 2.7%, contralateral invasive breast cancer 1.3%, non-breast second primary malignancies 2.3%, death 0%). A statistically significant improvement in DDFS in the olaparib arm compared with the placebo arm was also observed. At the next planned OS analysis, a statistically significant improvement in OS was observed in the olaparib arm compared with the placebo arm. Efficacy results in the FAS are presented in Table 11 and Figures 9 and 10.
Table 11. Efficacy results for adjuvant treatment of patients with germline BRCA-mutated early breast cancer in OlympiA:
| Olaparib 300 mg bd (N=921) | Placebo (N=915) | |
|---|---|---|
| IDFS (15% maturity) – DCO 27 March 2020 | ||
| Number of events: Total number of patients (%) | 106:921 (12) | 178:915 (20) |
| HR (99.5% CI)a | 0.58 (0.41, 0.82) | |
| P value (2-sided)b | 0.0000073 | |
| Percentage (95% CI) of patients invasive disease free at 3 yearsc | 86 (83, 88) | 77 (74, 80) |
| DDFS (13% maturity) – DCO 27 March 2020 | ||
| Number of events: Total number of patients (%) | 89:921 (10) | 152:915 (17) |
| HR (99.5% CI)a | 0.57 (0.39, 0.83) | |
| P value (2-sided)b | 0.0000257 | |
| Percentage (95% CI) of patients distant disease free at 3 yearsc | 88 (85, 90) | 80 (77, 83) |
| OS (10% maturity) – DCO 12 July 2021 | ||
| Number of events: Total number of patients (%) | 75:921 (8) | 109:915 (12) |
| HR (98.5% CI)a | 0.68 (0.47, 0.97) | |
| P value (2-sided)b | 0.0091 | |
| Percentage (95% CI) of patients alive at 3 yearsc | 93 (91, 94) | 89 (87, 91) |
| Percentage (95% CI) of patients alive at 4 yearsc | 90 (87, 92) | 86 (84, 89) |
a Based on the stratified Cox’s proportional hazards model, <1 indicates a lower risk with olaparib compared with placebo arm.
b P-value from a stratified log-rank test.
c Percentages are calculated using KM estimates.
bd = twice daily; CI = confidence interval; DDFS = distant disease free survival; IDFS = invasive disease free survival; KM = Kaplan-Meier; OS = overall survival.
Figure 9. Kaplan-Meier plot of IDFS for adjuvant treatment of patients with germline BRCA-mutated high risk early breast cancer in OlympiA:

Figure 10. Kaplan-Meier plot of OS for adjuvant treatment of patients with germline BRCA-mutated high risk early breast cancer in OlympiA:

gBRCA1/2-mutated HER2-negative metastatic breast cancer
OlympiAD (Study D0819C00003)
The safety and efficacy of olaparib in patients with gBRCA1/2-mutations who had HER2-negative metastatic breast cancer were studied in a Phase III randomised, open-label, controlled trial (OlympiAD). In this study 302 patients with a documented deleterious or suspected deleterious gBRCA mutation were randomised 2:1 to receive either Lynparza (300 mg [2 × 150 mg tablets] twice daily) or physician’s choice of chemotherapy (capecitabine 42%, eribulin 35%, or vinorelbine 17%) until progression or unacceptable toxicity. Patients with BRCA1/2 mutations were identified from germline testing in blood via a local test or by central testing at Myriad. Patients were stratified based on: receipt of prior chemotherapy regimens for metastatic breast cancer (yes/no), hormone receptor (HR) positive vs triple negative (TNBC), prior platinum treatment for breast cancer (yes/no). The primary endpoint was PFS assessed by blinded independent central review (BICR) using RECIST 1.1. Secondary endpoints included PFS2, OS, objective response rate (ORR) and HRQoL.
Patients must have received treatment with an anthracycline unless contraindicated and a taxane in either a (neo)adjuvant or metastatic setting. Patients with HR+ (ER and/or PgR positive) tumours must have received and progressed on at least one endocrine therapy (adjuvant or metastatic) or had disease that the treating physician believed to be inappropriate for endocrine therapy. Prior therapy with platinum was allowed in the metastatic setting provided there had been no evidence of disease progression during platinum treatment and in the (neo)adjuvant setting provided the last dose was received at least 12 months prior to randomisation. No previous treatment with a PARP inhibitor, including olaparib, was permitted.
Demographic and baseline characteristics were generally well balanced between the olaparib and comparator arms (see Table 12).
Table 12. Patient demographic and baseline characteristics in OlympiAD:
| Olaparib 300 mg bd n=205 | Chemotherapy n=97 | |
|---|---|---|
| Age – year (median) | 44 | 45 |
| Gender (%) | ||
| Female | 200 (98) | 95 (98) |
| Male | 5 (2) | 2 (2) |
| Race (%) | ||
| White | 134 (65) | 63 (65) |
| Asian | 66 (32) | 28 (29) |
| Other | 5 (2) | 6 (6) |
| ECOG performance status (%) | ||
| 0 | 148 (72) | 62 (64) |
| 1 | 57 (28) | 35 (36) |
| Overall disease classification | ||
| Metastatic | 205 (100) | 97 (100) |
| Locally advanced | 0 | 0 |
| New metastatic breast cancer (%) | 26 (13) | 12 (12) |
| Hormone receptor status (%) | ||
| HR+ | 103 (50) | 49 (51) |
| TNBC | 102 (50) | 48 (49) |
| gBRCA mutation type (%) | ||
| gBRCA1 | 117 (57) | 51 (53) |
| gBRCA2 | 84 (41) | 46 (47) |
| gBRCA1 and gBRCA2 | 4 (2) | 0 |
| ≥2 Metastatic sites (%) | 159 (78) | 72 (74) |
| Location of the metastasis (%) | ||
| Bone only | 16 (8) | 6 (6) |
| Other | 189 (92) | 91 (94) |
| Measurable disease by BICR (%) | 167 (81) | 66 (68) |
| Progressive disease at time of randomization (%) | 159 (78) | 73 (75) |
| Tumour grade at diagnosis | ||
| Well differentiated (G1) | 5 (2) | 2 (2) |
| Moderately differentiated (G2) | 52 (25) | 23 (24) |
| Poorly differentiated (G3) | 108 (53) | 55 (57) |
| Undifferentiated (G4) | 4 (2) | 0 |
| Unassessable (GX) | 27 (13) | 15 (16) |
| Missing | 9 (4) | 2 (2) |
| Number of prior lines of chemotherapy for metastatic breast cancer (%) | ||
| 0 | 68 (33) | 31 (32) |
| 1 | 80 (39) | 42 (43) |
| 2 | 57 (28) | 24 (25) |
| Previous platinum-based therapy (%) | 55 (27) | 21 (22) |
| in (neo)adjuvant setting only | 12 (6) | 6 (6) |
| metastatic setting only | 40 (20) | 14 (14) |
| in (neo)adjuvant and metastatic setting | 3 (1) | 1 (1) |
| Previous anthracycline treatment | ||
| in (neo)adjuvant setting | 169 (82) | 76 (78) |
| metastatic setting | 41 (20) | 16 (17) |
| Previous taxane treatment | ||
| in (neo)adjuvant setting | 146 (71) | 66 (68) |
| metastatic setting | 107 (52) | 41 (42) |
| Previous anthracycline and taxane treatment | 204 (99.5) | 96 (99) |
As subsequent therapy, 0.5% and 8% of patients received a PARP inhibitor in the treatment and comparator arms, respectively; 29% and 42% of patients, respectively, received subsequent platinum therapy.
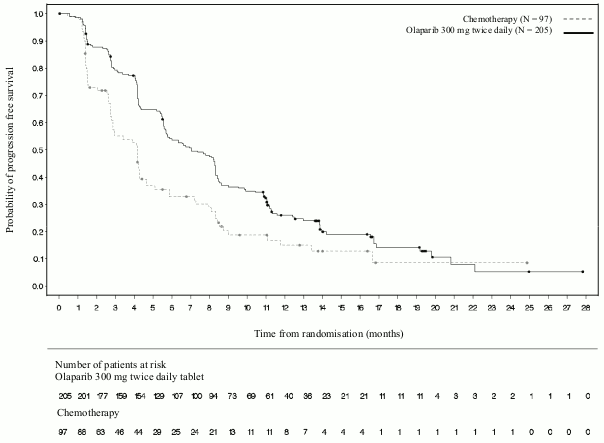
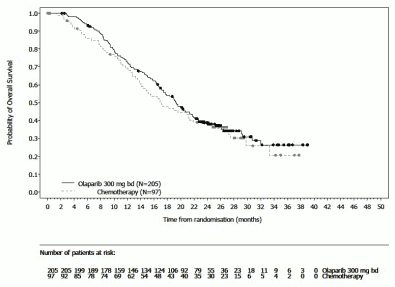
A statistically significant improvement in PFS, the primary efficacy outcome, was demonstrated for olaparib-treated patients compared with those in the comparator arm (see Table 13 and Figure 11).
Table 13. Summary of key efficacy findings for patients with gBRCA1/2-mutated HER2-negative metastatic breast cancer in OlympiAD:
| Olaparib 300 mg bd | Chemotherapy | |
|---|---|---|
| PFS (77% maturity) – DCO 09 December 2016 | ||
| Number of events: Total number of patients (%) | 163:205 (80) | 71:97 (73) |
| Median time (months) (95% CI) | 7.0 (5.7-8.3) | 4.2 (2.8-4.3) |
| HR (95% CI) | 0.58 (0.43-0.80) | |
| P value (2-sided)a | p=0.0009 | |
| PFS2 (65% maturity) - DCO 25 September 2017b | ||
| Number of events: Total number of patients (%) | 130:205 (63) | 65:97 (67) |
| Median time (months) (95% CI) | 12.8 (10.9-14.3) | 9.4 (7.4-10.3) |
| HR (95% CI) | 0.55 (0.39-0.77) | |
| P value (2-sided)a | p=0.0005 | |
| OS (64% maturity) – DCO 25 September 2017 | ||
| Number of events: Total number of patients (%) | 130:205 (63) | 62:97 (64) |
| Median time (months) (95% CI) | 19.3 (17.2-21.6)c | 17.1 (13.9-21.9) |
| HR (95% CI) | 0.90 (0.66-1.23) | |
| P value (2-sided)a | p=0.5131 | |
| Confirmed ORR – DCO 09 December 2016 | ||
| Number of objective responders: Total number of patients with measurable disease (%) | 87: 167 (52)d | 15:66 (23) |
| 95% CI | 44.2-59.9 | 13.3-35.7 |
| DOR – DCO 09 December 2016 | ||
| Median, months (95% CI) | 6.9 (4.2, 10.2) | 7.9 (4.5, 12.2) |
a Based on stratified log-rank test.
b Post-hoc analysis.
c The median follow-up time in censored patients was 25.3 months for olaparib versus 26.3 months for comparator.
d Confirmed responses (by BICR) were defined as a recorded response of either CR/PR, confirmed by repeat imaging not less than 4 weeks after the visit when the response was first observed. In the olaparib arm 8% with measurable disease had a complete response versus 1.5% of patients in the comparator arm; 74/167 (44%) of patients in the olaparib arm had a partial response versus 14/66 (21%) of patients in the chemotherapy arm. In the TNBC patient subgroup the confirmed ORR was 48% (41/86) in the olaparib arm and 12% (4/33) in the comparator arm. In the HR+ patient subgroup the confirmed ORR was 57% (46/81) in the olaparib arm and 33% (11/33) in the comparator arm.
bd Twice daily; CI Confidence interval; DOR Duration of response; DCO Data cut off; HR Hazard ratio; HR+ Hormone receptor positive, ORR Objective response rate; OS overall survival; PFS progression-free survival; PFS2 Time to second progression or death, TNBC triple negative breast cancer.
Figure 11. OlympiAD: Kaplan-Meier plot of BICR PFS in patients with gBRCA1/2-mutated HER2-negative metastatic breast cancer (77% maturity) DCO 09 December 2016:

Consistent results were observed in all predefined patient subgroups (see Figure 12). Subgroup analysis indicated PFS benefit of olaparib versus comparator in TNBC (HR 0.43; 95% CI: 0.29-0.63, n=152) and HR+ (HR 0.82; 95% CI: 0.55-1.26, n=150) patient subgroups.
Figure 12. PFS (BICR), Forest plot, by prespecified subgroup:

In a post-hoc analysis of the subgroup of patients that had not progressed on chemotherapy other than platinum, the median PFS in the olaparib arm (n=22) was 8.3 months (95% CI 3.1-16.7) and 2.8 months (95% CI 1.4-4.2) in the chemotherapy arm (n=16) with a HR of 0.54 (95% CI 0.24-1.23). However, the number of patients is too limited to make meaningful conclusions on the efficacy in this subgroup.
Seven male patients were randomised (5 olaparib and 2 comparator). At the time of the PFS analysis, 1 patient had a confirmed partial response with a duration of response of 9.7 months in the olaparib arm. There were no confirmed responses in the comparator arm.
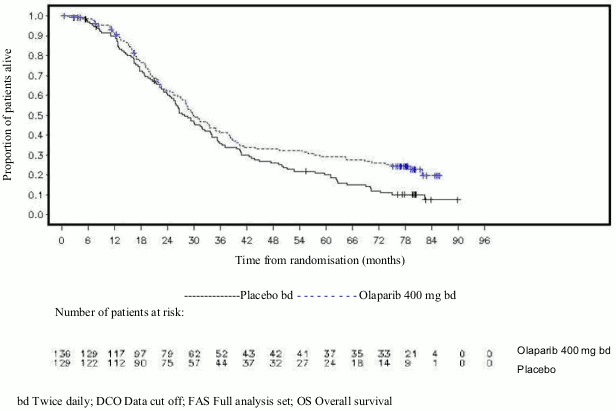
Figure 13. OlympiAD: Kaplan-Meier plot of OS in patients with gBRCA1/2-mutated HER2-negative metastatic breast cancer (64% maturity) DCO 25 September 2017:

OS analysis in patients with no prior chemotherapy for metastatic breast cancer indicated benefit in these patients with a HR of 0.45 (95% CI 0.27-0.77), while for further lines of therapy HR exceeded 1.
Maintenance following first-line treatment of germline BRCA-mutated metastatic adenocarcinoma of the pancreas
POLO Study
The safety and efficacy of olaparib as maintenance therapy were studied in a randomised (3:2), double-blind, placebo-controlled, multicentre trial in 154 patients with germline BRCA1/2 mutations who had metastatic adenocarcinoma of the pancreas. Patients received either Lynparza 300 mg (2 × 150 mg tablets) twice daily (n=92) or placebo (n=62) until radiological disease progression or unacceptable toxicity. Patients should have not progressed during first-line platinum-based chemotherapy and should have received a minimum of 16 weeks of continuous platinum treatment, which could be discontinued at any time thereafter for unacceptable toxicity while the remaining agents continued according to the planned regimen or unacceptable toxicity for other component(s). Patients who could tolerate complete platinum-containing chemotherapy regimen until progression have not been considered for this study. The maintenance therapy was started 4 to 8 weeks after the last dose of first-line chemotherapy component(s) in the absence of progression and if all toxicities from previous anti-cancer therapy had been resolved to CTCAE grade 1, except for alopecia, grade 3 peripheral neuropathy and Hgb ≥9 g/dL.
Thirty-one percent (31%) of patients with germline BRCA1/2 mutations were identified from prior local testing results and 69% of patients by central testing. In the olaparib arm, 32% of patients carried a germline BRCA1 mutation, 64% a germline BRCA2 mutation and 1% carried both germline BRCA1 and germline BRCA2 mutations. In the placebo arm, 26% of patients carried a germline BRCA1 mutation, 73% a germline BRCA2 mutation and no patients carried both germline BRCA1 and germline BRCA2 mutations. The BRCAm status of all patients identified using prior local testing results was confirmed, where sent, by central testing. Ninety-eight percent (98%) of patients carried a deleterious mutation and 2% carried a suspected deleterious mutation. Large rearrangements in the BRCA1/2 genes were detected in 5.2 % (8/154) of the randomised patients.
Demographic and baseline characteristics were generally well balanced between the olaparib and placebo arms. Median age was 57 years in both arms; 30% of patients in the olaparib arm were ≥65 years compared to 20% in the placebo arm. Fifty-eight per-cent (58%) of patients in the olaparib arm and 50% of patients in the placebo arm were male. In the olaparib arm 89% of patients were White and 11% were non-White; in the placebo arm 95% of patients were White and 5% were non-White. Most patients were ECOG performance status 0 (71% in the olaparib arm and 61% in the placebo arm). Overall, the sites of metastasis prior to chemotherapy were liver 72%, lung 10% and other sites 50%. The median time from original diagnosis to randomisation across both arms was 6.9 months (range 3.6 to 38.4 months).
Overall, 75% of patients received FOLFIRINOX with a median of 9 cycles (range 4-61), 8% received FOLFOX or XELOX, 4% received GEMOX, and 3% received gemcitabine plus cisplatin; the remaining 10% of patients received other chemotherapy regimens. Duration of the first-line chemotherapy for metastatic disease was 4 to 6 months, >6 to <12 months and ≥12 months, respectively, in 77%, 19% and 4% of patients in the olaparib arm and in 80%, 17% and 3% in the placebo arm, with around 1 month from the last dose of the first-line chemotherapy component(s) to the start of study treatment in both arms. As best response on first-line chemotherapy, 7% of olaparib patients and 5% of placebo patients had a complete response, 44% of olaparib patients and 44% of placebo patients had a partial response and 49% of olaparib and 50% of placebo patients had stable disease. At randomisation, measurable disease was reported in 85% and 84% of patients in the olaparib or placebo arms, respectively. The median time from initiation of the first-line platinum- based chemotherapy to randomisation was 5.7 months (range 3.4 to 33.4 months).
At the time of PFS analysis, 33% of patients in the olaparib arm and 13% on the placebo arm remained on study treatment. Forty-nine percent of patients (49%) in the olaparib arm and 74% in the placebo arm received subsequent therapy. Forty-two percent (42%) of patients in the olaparib arm and 55% in the placebo arm received platinum as subsequent therapy. One percent (1%) of patients in the olaparib arm and 15% in the placebo arm received PARP inhibitor as subsequent therapy. Of the 33 (36%) and 28 (45%) of patients who received a first subsequent platinum-containing therapy, in the olaparib and placebo arms, stable disease was reported in 8 vs 6 patients, whereas 1 vs 2 patients had responses, respectively.
The primary endpoint was progression-free survival (PFS), defined as time from randomisation to progression determined by BICR using Response Evaluation Criteria in Solid Tumors (RECIST) 1.1 modified to assess patients with no evidence of disease, or death. Secondary efficacy endpoints included overall survival (OS), time from randomisation to second progression or death (PFS2), time from randomisation to first subsequent anti-cancer therapy or death (TFST), objective response rate (ORR), duration of response (DoR), response rate, time to response and health related quality of life (HRQoL).
The study demonstrated a statistically significant improvement in PFS for olaparib compared to placebo (Table 14). The BICR assessment of PFS was consistent with an investigator assessment.
At final analysis of OS, the percentage of patients that were alive and in follow-up was 28% in the olaparib arm and 18% in the placebo arm.
Table 14. Efficacy results for patients with gBRCAm metastatic adenocarcinoma of the pancreas in POLO:
| Olaparib 300 mg bd | Placebo | |
|---|---|---|
| PFS (68% maturity)a,b (BICR, DCO 15 January 2019) | ||
| Number of events: Total number of patients (%) | 60:92 (65) | 44:62 (71) |
| Median time, months (95% CI) | 7.4 (4.14-11.01) | <3.8 (3.52-4.86) |
| HR (95% CI)c,d | 0.53 (0.35-0.82) | |
| P value (2-sided) | p=0.0038 | |
| OS (70% maturity)e (DCO 21 July 2020) | ||
| Number of events: Total number of patients (%) | 61:92 (66) | 47:62 (76) |
| Median time (months) (95% CI) | 19.0 (15.28-26.32) | <19.2 (14.32-26.12) |
| HR (95% CI)d | 0.83 (0.56-1.22) | |
| P value (2-sided) | p=0.3487 | |
a Based on Kaplan–Meier estimates, the proportion of patients that were alive and progression-free at 12 and 24 months were 34% and 22% for olaparib vs 15% and 10% for placebo.
b For PFS, the median follow-up time for censored patients was 9.1 months in the olaparib arm and 3.8 months in the placebo arm.
c A value <1 favours olaparib.
d The analysis was performed using a log-rank test.
e For OS, the median follow-up time for censored patients was 31.3 months in the olaparib arm and 23.9 months in the placebo arm.
bd Twice daily; CI Confidence interval; HR Hazard Ratio; OS Overall Survival; PFS Progression-free survival.
Figure 14. POLO: Kaplan-Meier plot of PFS for patients with gBRCAm metastatic adenocarcinoma of the pancreas (68% maturity – BICR, DCO 15 January 2019):

Figure 15. POLO: Kaplan-Meier plot of OS for patients with gBRCAm metastatic adenocarcinoma of the pancreas (70% maturity, DCO 21 July 2020):

BRCA1/2-mutated metastatic castration-resistant prostate cancer:
PROfound Study
The safety and efficacy of olaparib were studied in men with metastatic castration-resistant prostate cancer (mCRPC) in a Phase III randomised, open-label, multicentre trial that evaluated the efficacy of Lynparza versus a comparator arm of investigator’s choice of NHA ([new hormonal agent] enzalutamide or abiraterone acetate).
Patients needed to have progressed on prior NHA for the treatment of metastatic prostate cancer and/or CRPC. For inclusion in Cohort A, patients needed to have deleterious or suspected deleterious mutations in either BRCA1 or BRCA2 genes. Patients with ATM mutations were also randomised in Cohort A, but positive benefit-risk could not be demonstrated in this subpopulation of patients. Patients with mutations in other genes were randomised in Cohort B.
In this study 387 patients were randomised 2:1 to receive either olaparib (300 mg [2 × 150 mg tablets] twice daily) or comparator. In Cohort A there were 245 patients (162 olaparib and 83 comparator) and in Cohort B there were 142 patients (94 olaparib and 48 comparator). Patients were stratified by prior taxane use and evidence of measurable disease. Treatment was continued until disease progression. Patients randomised to comparator were given the option to switch to olaparib upon confirmed radiological BICR progression. Patients with BRCA1m, BRCA2m detected in their tumours were enrolled on the basis of prospective central testing, with the exception of 3 patients enrolled using a local test result. Of the 160 patients with a BRCA1 or BRCA2 mutation in PROfound, 114 patients were retrospectively tested to determine if the identified BRCA1/2 mutation was germline or somatic in origin. Within these patients, 63 BRCA1/2 mutations were identified in the germline blood sample and hence were determined to be germline in origin. The remaining 51 patients did not have a tumour detected BRCA1/2 mutation identified in the germline blood sample and hence the BRCA1/2 mutations are determined to be somatic in origin. For the remaining 46 patients, somatic or germline origin is unknown.
Demographics and baseline characteristics were generally well balanced between the olaparib and comparator arms in patients with BRCA1/2 mutations. Median age was 68 years and 67 years in the olaparib and comparator arms, respectively. Prior therapy in the olaparib arm was 71% taxane, 41% enzalutamide, 37% abiraterone acetate and 20% both enzalutamide and abiraterone acetate. Prior therapy in the comparator arm was 60% taxane, 50% enzalutamide, 36% abiraterone acetate and 14% both enzalutamide and abiraterone acetate. Fifty-eight percent (58%) of patients in the olaparib arm and 55% in the comparator arm had measurable disease at study entry. The proportion of patients with bone, lymph node, respiratory and liver metastases was 89%, 62%, 23% and 12%, respectively in the olaparib arm and 86%, 71%, 16% and 17%, respectively in the comparator arm. Most patients in both treatment arms had an ECOG of 0 or 1 (93%). Baseline pain scores (BPI-SF worst pain) were 0-<2 (52%), 2-3 (10%) or >3 (34%) in the olaparib arm and 0-<2 (45%), 2-3 (7%) or >3 (45%) in the comparator arm. Median baseline PSA was 57.48 μg/L in the olaparib arm and 103.95 μg/L in the comparator.
The primary endpoint of the study was radiological progression free survival (rPFS) in Cohort A determined by BICR using RECIST 1.1 (soft tissue) and Prostate Cancer Working Group (PCWG3) (bone). Key secondary endpoints included confirmed objective response rate (ORR) by BICR, rPFS by BICR, time to pain progression (TTPP) and overall survival (OS).
The study demonstrated a statistically significant improvement in BICR assessed rPFS and final OS for olaparib vs comparator in Cohort A.
Results for patients with BRCA1/2 mutations are presented in Table 15. There was a statistically significant improvement in BICR assessed rPFS for olaparib vs the investigators choice of NHA arm in BRCA1/2m patients. The final analysis of OS showed a nominally statistically significant improvement in OS in BRCA1/2m patients randomised to Lynparza vs comparator.
Table 15. Summary of key efficacy findings in patients with BRCA1/2-mutated mCRPC in PROfound:
| Olaparib 300 mg bd (N=102) | Investigators choice of NHA (N=58) | |
|---|---|---|
| rPFS by BICRa,b,c DCO 4 June 2019 | ||
| Number of events: Total number of patients (%) | 62:102 (61)c | 51:58 (88)c |
| Median rPFS (95% CI) [months] | 9.8 (7.6, 11.3) | 3.0 (1.8, 3.6) |
| HR (95% CI)c | 0.22 (0.15, 0.32) | |
| Confirmed ORR by BICRa | ||
| Number of objective responders: Total number of patients with measurable disease at baseline (%) | 25:57 (44) | 0:33 (0) |
| Odds ratio (95% CI) | NC (NC, NC) | |
| OSa DCO 20 March 2020c | ||
| Number of events: Total number of patients (%) | 53:102 (52) | 41:58 (71) |
| Median OS (95% CI) [months] | 20.1 (17.4, 26.8) | 14.4 (10.7, 18.9) |
| HR (95% CI) | 0.63 (0.42, 0.95) | |
a Not controlled for multiplicity
b rPFS 71% maturity
c The HR and CI were calculated using a Cox proportional hazards model that contains terms for treatment, factor and treatment by factor interaction.
bd Twice daily; BICR Blinded independent central review; CI Confidence interval; HR Hazard ratio; NC Not calculable; NHA New hormonal agent; ORR Objective response rate; OS Overall survival; rPFS Radiological progression-free survival
Figure 16. BRCA1/2m patients: Kaplan-Meier plot of rPFS (by BICR):

Figure 17. BRCA1/2m patients: Kaplan-Meier plot of OS:

Treatment of patients in the first-line mCRPC setting
PROpel
The safety and efficacy of olaparib were studied in men with metastatic castration-resistant prostate cancer (mCRPC) in a Phase III randomised, double-blind, placebo-controlled, multicentre study that evaluated the efficacy of Lynparza (300 mg [2 × 150 mg tablets] twice daily) in combination with abiraterone (1000 mg [2 × 500 mg tablets] once daily) versus a comparator arm of placebo plus abiraterone. Patients in both arms also received either prednisone or prednisolone 5 mg twice daily.
The study randomised 796 patients (1:1 randomisation; 399 olaparib/abiraterone:397 placebo/ abiraterone) who had evidence of histologically confirmed prostate adenocarcinoma and metastatic status defined as at least one documented metastatic lesion on either a bone or CT/MRI scan and who were treatment naïve with no prior chemotherapy or NHA in the mCRPC setting. Prior to the mCRPC stage, treatment with NHAs (except abiraterone) without PSA progression (clinical or radiological) during treatment was allowed, provided the treatment was stopped at least 12 months before randomisation. Treatment with first-generation antiandrogen agents (e.g., bicalutamide, nilutamide, flutamide) was also allowed, provided there was a washout period of 4 weeks. Docetaxel treatment was allowed during neoadjuvant/adjuvant treatment for localised prostate cancer and at metastatic hormone-sensitive prostate cancer (mHSPC) stage, as long as no signs of disease progression occurred during or immediately after such treatment. All patients received a GnRH analogue or had prior bilateral orchiectomy. Patients were stratified by metastases (bone only, visceral or other) and docetaxel treatment at mHSPC stage (yes or no). Treatment was continued until radiological progression of the underlying disease or unacceptable toxicity.
Demographic and baseline characteristics were balanced between the two treatment arms. The median age of patients was 69 years overall, and the majority (71%) of patients were in the ≥65 years age group. One hundred and eighty-nine patients (24%) had prior docetaxel treatment at mHSPC stage. In total, 434 (55%) patients had bone metastases (metastases in the bone and no other distant site), 105 (13%) patients had visceral metastases (distant soft tissue metastases in an organ e.g., liver, lung) and 257 (32%) patients had other metastases (this could include, for example, patients with bone metastases and distant lymph nodes or patients with disease present only in distant lymph nodes). Most patients in both arms (70%) had an ECOG performance status of 0. There were 103 (25.8%) symptomatic patients in the olaparib group and 80 (20.2%) patients in the placebo group. Symptomatic patients were characterized by Brief Pain Inventory-Short Form (BPI-SF) item #3 score ≥ 4 and/or opiate use at baseline.
Patient enrolment was not based on biomarker status. HRR gene mutation status was assessed retrospectively by ctDNA and tumour tissue tests to assess the consistency of treatment effect from the FAS population. Of the patients tested, 198 and 118 were HRRm as determined by ctDNA and tumour tissue, respectively. The distribution of HRRm patients was well balanced between the two arms.
The primary endpoint was rPFS, defined as time from randomisation to radiological progression determined by investigator assessment based on RECIST 1.1 and PCWG-3 criteria (bone). The key secondary efficacy endpoint was overall survival (OS). Additional secondary endpoints included PFS2, TFST and HRQoL.
The study met its primary endpoint demonstrating a statistically significant improvement in the risk of radiological disease progression or death for olaparib/abiraterone compared to placebo/abiraterone as assessed by the investigator, with HR 0.66; 95% CI 0.54, 0.81; p<0.0001; median rPFS 24.8 months in the olaparib/abiraterone arm vs 16.6 months in the placebo/abiraterone arm. The investigator assessment of rPFS was supported with a blinded independent central radiological (BICR) review. The sensitivity analysis of rPFS by BICR was consistent with the investigator-based analysis with HR 0.61; 95% CI 0.49, 0.74; p<0.0001; median rPFS 27.6 months in the olaparib/abiraterone arm vs 16.4 months in the placebo/abiraterone arm, respectively.
Subgroup results were consistent with the overall results for olaparib/abiraterone compared to placebo/abiraterone in all pre-defined sub-groups, including patients with or without prior taxane at mHSPC stage, patients with different metastatic disease at baseline (bone only vs visceral vs other) and patients with or without HRRm (Figure 20).
Efficacy results are presented in Table 16, Table 17, Figure 18 and Figure 19.
Table 16. Summary of key efficacy findings for treatment of patients with mCRPC in PROpel:
| Olaparib/abiraterone N=399 | Placebo/abiraterone N=397 | |
|---|---|---|
| rPFS (by investigator assessment) (50% maturity) (DCO 30 July 2021) | ||
| Number of events: Total number of patients (%) | 168:399 (42.1) | 226:397 (56.9) |
| Median time (95% CI) (months) | 24.8 (20.5, 27.6) | 16.6 (13.9, 19.2) |
| HR (95% CI)a | 0.66 (0.54, 0.81) | |
| p-valueb | <0.0001 | |
| Final OS (48% maturity) (DCO 12 October 2022) | ||
| Number of events: Total number of patients (%) | 176:399 (44.1) | 205:397 (51.6) |
| Median time (95% CI) (months) | 42.1 (38.4, NC) | 34.7 (31.0, 39.3) |
| HR (95% CI)a | 0.81 (0.67, 1.00) | |
| p-valueb | p=0.0544 | |
| % Alive at 36 months (95% CI)c | 56.9 (51.7, 61.7) | 49.5 (44.3, 54.5) |
a The HR and CI were calculated using a Cox proportional hazards model adjusted for the variables selected in the primary pooling strategy: metastases, docetaxel treatment at mHSPC stage. The Efron approach was used for handling ties. A HR <1 favours olaparib 300 mg bd + abiraterone 1000 mg qd.
b The 2-sided p-value was calculated using the log-rank test stratified by the same variables selected in the primary pooling strategy.
c Calculated using the Kaplan-Meier technique.
Table 17. rPFS subgroup analyses by investigator assessment in PROpel (DCO 30 July 2021):
| Olaparib/abiraterone | Placebo/abiraterone | |
|---|---|---|
| Radiological Progression-Free Survival (rPFS) by investigator assessment | ||
| Aggregate HRRm Subgroup Analysesa | ||
| HRRm | N=111 | N=115 |
| Number of events: Total number of patients (%) | 43:111 (38.7) | 73:115 (63.5) |
| Median (months) | NC | 13.86 |
| Hazard ratio (95% CI)b | 0.50 (0.34, 0.73) | |
| Non-HRRm | N=279 | N=273 |
| Number of events: Total number of patients (%) | 119:279 (42.7) | 149:273 (54.6) |
| Median (months) | 24.11 | 18.96 |
| Hazard ratio (95% CI)b | 0.76 (0.60, 0.97) | |
| Aggregate BRCAm Subgroup Analysesa | ||
| BRCAm | N=47 | N=38 |
| Number of events: Total number of patients (%) | 14:47 (29.8) | 28:38 (73.7) |
| Median (months) | NC | 8.38 |
| Hazard ratio (95% CI)b | 0.23 (0.12, 0.43) | |
| Non-BRCAm | N=343 | N=350 |
| Number of events: Total number of patients (%) | 148:343 (43.1) | 194:350 (55.4) |
| Median (months) | 24.11 | 18.96 |
| Hazard ratio (95% CI)b | 0.76 (0.61, 0.94) | |
a Aggregate subgroups were derived from ctDNA and tissue-based groupings.
b The analysis was performed using a Cox proportional hazards model including terms for treatment group, the subgroup factor, and a treatment by subgro
Figure 18. PROpel: Kaplan-Meier plot of rPFS (investigator assessed) (50% maturity) DCO 30 July 2021:

Figure 19. PROpel: Kaplan-Meier plot of OS (48% maturity) DCO 12 October 2022:

Figure 20. PROpel: Forest plot of subgroup analysis of rPFS (investigator assessed) (50% maturity) DCO 30 July 2021:

Each subgroup analysis was performed using a Cox proportional hazards model that contained a term for treatment, factor, and treatment by factor interaction. A hazard ratio <1 implies a lower risk of progression on olaparib. The size of a circle is proportional to the number of events. All subgroups in this figure are based upon data from the eCRF.
* Excludes patients with no baseline assessment. CI: confidence interval, ECOG: Eastern Cooperative Oncology Group; HRRm: homologous recombination repair gene mutation; mHSPC: metastatic hormone-sensitive prostate cancer; NC: noncalculable; PSA: prostate-specific antigen
First-line maintenance treatment of mismatch repair proficient (pMMR) advanced or recurrent endometrial cancer
DUO-E Study
DUO-E was a randomised, multicentre, double-blind, placebo-controlled, Phase III study of first-line platinum-based chemotherapy in combination with durvalumab, followed by durvalumab with or without olaparib in patients with advanced or recurrent endometrial cancer. Patients had to have endometrial cancer in one of the following categories: newly diagnosed Stage III disease (measurable disease per RECIST 1.1 following surgery or diagnostic biopsy), newly diagnosed Stage IV disease (with or without disease following surgery or diagnostic biopsy), or recurrence of disease (measurable or non-measurable disease per RECIST 1.1) where the potential for cure by surgery alone or in combination is poor. For patients with recurrent disease, prior chemotherapy was allowed only if it was administered in the adjuvant setting and there was at least 12 months from the date of last dose of chemotherapy administered to the date of subsequent relapse. The study included patients with epithelial endometrial carcinomas of all histologies, including carcinosarcomas. Patients with endometrial sarcoma were excluded.
Randomisation was stratified by tumour tissue’s mismatch repair (MMR) status (proficient versus deficient), disease status (recurrent versus newly diagnosed), and geographic region (Asia versus rest of the world). Patients were randomised 1:1:1 to one of the following arms:
- Platinum-based chemotherapy: Platinum-based chemotherapy (paclitaxel and carboplatin) every 3 weeks for a maximum of 6 cycles with durvalumab placebo every 3 weeks. Following completion of chemotherapy treatment, patients without objective disease progression received durvalumab placebo every 4 weeks and olaparib placebo tablets twice daily as maintenance treatment until disease progression.
- Platinum-based chemotherapy + durvalumab: Platinum-based chemotherapy (paclitaxel and carboplatin) every 3 weeks for a maximum of 6 cycles with 1120 mg durvalumab every 3 weeks. Following completion of chemotherapy treatment, patients without objective disease progression received 1500 mg durvalumab every 4 weeks with olaparib placebo tablets twice daily as maintenance treatment until disease progression.
- Platinum-based chemotherapy + durvalumab + olaparib: Platinum-based chemotherapy (paclitaxel and carboplatin) every 3 weeks for a maximum of 6 cycles with 1120 mg durvalumab every 3 weeks. Following completion of chemotherapy treatment, patients without objective disease progression received 1500 mg durvalumab every 4 weeks with 300 mg olaparib tablets twice daily as maintenance treatment until disease progression.
Patients who discontinued either product (olaparib/placebo or durvalumab/placebo) for reasons other than disease progression could continue treatment with the other product if appropriate based on toxicity considerations and investigator discretion.
Treatment was continued until RECIST v1.1-defined progression of disease or unacceptable toxicity. Assessment of tumour status was performed every 9 weeks for the first 18 weeks relative to randomisation and every 12 weeks thereafter.
The primary endpoint was PFS, defined as time from randomisation to progression determined by investigator assessment using RECIST 1.1, or death. Secondary efficacy endpoints included OS, ORR and DoR.
The study demonstrated a statistically significant improvement in PFS in the ITT population for patients treated with platinum-based chemotherapy + durvalumab + olaparib compared to platinum- based chemotherapy alone (HR 0.55; 95% CI: 0.43, 0.69). At the time of PFS analysis, interim OS data were 28% mature with events in 199 of 718 patients.
Mismatch repair (MMR) status was determined centrally using an MMR immunohistochemistry panel assay. Of a total of 718 patients randomized in the study, 575 (80%) patients had MMR-proficient (pMMR) tumour status and 143 (20%) patients had MMR-deficient (dMMR) tumour status.
Among patients with pMMR tumour status, demographic and baseline characteristics were generally well balanced between the treatment arms. Baseline demographics across all three arms were as follows: median age of 64 years (range: 22 to 86); 48% age 65 or older; 8% age 75 or older; 56% White, 30% Asian, and 6% Black or African American. Disease characteristics were as follows: ECOG PS of 0 (69%) or 1 (31%); 47% newly diagnosed and 53% recurrent disease. The histologic subtypes were endometrioid (54%), serous (26%), carcinosarcoma (8%), mixed epithelial (4%), clear cell (3%), undifferentiated (2%), mucinous (<1%), and other (3%).
Results in patients with pMMR tumour status are summarised in Table 18 and Figure 21. The median follow-up time in censored patients with pMMR tumour status was 15.2 months in the platinum-based chemotherapy + durvalumab + olaparib arm and 12.8 months in the platinum-based chemotherapy arm. At the time of PFS analysis, interim OS data were 29% mature with events in 110 of 383 patients.
Table 18. Summary of efficacy results in patients with advanced or recurrent endometrial cancer in DUO-E (Patients with pMMR tumour status):
| Platinum-based chemotherapy + durvalumab + olaparib N=191 | Platinum-based chemotherapy N=192 | |
|---|---|---|
| PFS (by investigator assessment) (DCO 12 April 2023) | ||
| Number of events: Total number of patients (%) | 108:191 (56.5) | 148:192 (77.1) |
| Mediana (95% CI), months | 15.0 (12.4, 18.0) | 9.7 (9.2, 10.1) |
| HR (95% CI) | 0.57 (0.44, 0.73) | |
| OSb (DCO 12 April 2023) | ||
| Number of events: Total number of patients (%) | 46:191 (24.1) | 64:192 (33.3) |
| Mediana (95% CI), months | NR (NR, NR) | 25.9 (25.1, NR) |
| HR (95% CI) | 0.69 (0.47, 1.00) | |
| Objective response ratec (DCO 12 April 2023) | ||
| Number of objective responders: Total number of patients with measurable disease at baseline (%) | 90:147 (61.2) | 92:156 (59.0) |
| Duration of Response (DCO 12 April 2023) | ||
| Mediana (95% CI), months | 18.7 (10.5, NR) | 7.6 (7.1, 10.2) |
a Calculated using the Kaplan-Meier technique
b Based on first interim analysis
c Response: Best objective response as confirmed complete response or partial response.
CI Confidence interval; DCO Data cutoff; HR Hazard ratio; NR Not reached; OS Overall survival; PFS Progression-free survival
Figure 21. DUO-E: Kaplan-Meier plot of PFS (Patients with pMMR tumour status):

Among patients with pMMR tumour status, the PFS HRs were 0.44 (95% CI: 0.31, 0.61) in patients with PD-L1 expression positive status (236/383; 62%) and 0.87 (95% CI: 0.59, 1.28) in patients with PD-L1 expression negative status (140/383; 37%), for the platinum-based chemotherapy + durvalumab + olaparib arm vs the platinum-based chemotherapy arm. PD-L1 expression positive was defined as tumour area positive (TAP) ≥1%.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with Lynparza in all subsets of the paediatric population, in ovarian carcinoma (excluding rhabdomyosarcoma and germ cell tumours) (see section 4.2 for information on paediatric use).
Pharmacokinetic properties
The pharmacokinetics of olaparib at the 300 mg tablet dose are characterised by an apparent plasma clearance of ~7 L/h, an apparent volume of distribution of ~158 L and a terminal half-life of 15 hours. On multiple dosing, an AUC accumulation ratio of 1.8 was observed and PK appeared to be time-dependent to a small extent.
Absorption
Following oral administration of olaparib via the tablet formulation (2 × 150 mg), absorption is rapid with median peak plasma concentrations typically achieved 1.5 hours after dosing.
Co-administration with food slowed the rate (tmax delayed by 2.5 hours and Cmax reduced by approximately 21%) but did not significantly affect the extent of absorption of olaparib (AUC increased 8%). Consequently, Lynparza may be taken without regard to food (see section 4.2).
Distribution
The in vitro plasma protein binding is approximately 82% at 10 μg/mL which is approximately Cmax.
In vitro, human plasma protein binding of olaparib was dose-dependent; the fraction bound was approximately 91% at 1 μg/mL, reducing to 82% at 10 μg/mL and to 70% at 40 μg/mL. In solutions of purified proteins, the olaparib fraction bound to albumin was approximately 56%, which was independent of olaparib concentrations. Using the same assay, the fraction bound to alpha-1 acid glycoprotein was 29% at 10 μg/mL with a trend of decreased binding at higher concentrations.
Biotransformation
In vitro, CYP3A4/5 were shown to be the enzymes primarily responsible for the metabolism of olaparib (see section 4.5).
Following oral dosing of 14C-olaparib to female patients, unchanged olaparib accounted for the majority of the circulating radioactivity in plasma (70%) and was the major component found in both urine and faeces (15% and 6% of the dose, respectively). The metabolism of olaparib is extensive. The majority of the metabolism was attributable to oxidation reactions with a number of the components produced undergoing subsequent glucuronide or sulfate conjugation. Up to 20, 37 and 20 metabolites were detected in plasma, urine and faeces, respectively, the majority of them representing <1% of the dosed material. A ring-opened piperazin-3-ol moiety, and two mono-oxygenated metabolites (each ~10%) were the major circulating components, with one of the mono-oxygenated metabolites also being the major metabolite in the excreta (6% and 5% of the urinary and faecal radioactivity, respectively).
In vitro, olaparib produced little/no inhibition of UGT1A4, UGT1A9, UGT2B7, or CYPs 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6 or 2E1 and is not expected to be a clinically significant time dependent inhibitor of any of these CYP enzymes. Olaparib inhibited UGT1A1 in vitro, however, PBPK simulations suggest this is not of clinical importance. In vitro, olaparib is a substrate of the efflux transporter P-gp, however, this is unlikely to be of clinical significance (see section 4.5).
In vitro, data also show that olaparib is not a substrate for OATP1B1, OATP1B3, OCT1, BCRP or MRP2 and is not an inhibitor of OATP1B3, OAT1 or MRP2.
Elimination
Following a single dose of 14C-olaparib, ~86% of the dosed radioactivity was recovered within a 7-day collection period, ~44% via the urine and ~42% via the faeces. Majority of the material was excreted as metabolites.
Special populations
In population based PK analyses, patient age, gender, bodyweight, tumour location or race (including White and Japanese patients) were not significant covariates.
Renal impairment
In patients with mild renal impairment (creatinine clearance 51 to 80 ml/min), AUC increased by 24% and Cmax by 15% compared with patients with normal renal function. No Lynparza dose adjustment is required for patients with mild renal impairment.
In patients with moderate renal impairment (creatinine clearance 31 to 50 ml/min), AUC increased by 44% and Cmax by 26% compared with patients with normal renal function. Lynparza dose adjustment is recommended for patients with moderate renal impairment (see section 4.2).
There are no data in patients with severe renal impairment or end-stage renal disease (creatinine clearance <30 ml/min).
Hepatic impairment
In patients with mild hepatic impairment (Child-Pugh classification A), AUC increased by 15% and Cmax by 13% and in patients with moderate hepatic impairment (Child-Pugh classification B), AUC increased by 8% and Cmax decreased by 13% compared with patients with normal hepatic function. No Lynparza dose adjustment is required for patients with mild or moderate hepatic impairment (see section 4.2). There are no data in patients with severe hepatic impairment (Child-Pugh classification C).
Paediatric population
No studies have been conducted to investigate the pharmacokinetics of olaparib in paediatric patients.
Preclinical safety data
Repeat-dose toxicity
In repeat-dose toxicity studies of up to 6 months duration in rats and dogs, daily oral doses of olaparib were well-tolerated. The major primary target organ for toxicity in both species was the bone marrow, with associated changes in peripheral haematology parameters. These changes were reversible within 4 weeks of cessation of dosing. In rats, minimal degenerative effects on gastrointestinal tract were also noted. These findings occurred at exposures below those seen clinically. Studies using human bone marrow cells also showed that direct exposure to olaparib can result in toxicity to bone marrow cells in ex vivo assays.
Genotoxicity
Olaparib showed no mutagenic potential, but was clastogenic in mammalian cells in vitro. When dosed orally to rats, olaparib induced micronuclei in bone marrow. This clastogenicity is consistent with the known pharmacology of olaparib and indicates potential for genotoxicity in man.
Carcinogenicity
Carcinogenicity studies have not been conducted with olaparib.
Reproductive toxicology
In a female fertility study where rats were dosed until implantation, although extended oestrus was observed in some animals, mating performance and pregnancy rate was not affected. However, there was a slight reduction in embryofoetal survival.
In rat embryofoetal development studies, and at dose levels that did not induce significant maternal toxicity, olaparib caused reduced embryofoetal survival, reduced foetal weight and foetal developmental abnormalities, including major eye malformations (e.g. anophthalmia, microphthalmia), vertebral/rib malformation and visceral and skeletal abnormalities.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.