MULPLEO Film-coated tablet Ref.[7587] Active ingredients: Lusutrombopag
Source: European Medicines Agency (EU) Revision Year: 2021 Publisher: Shionogi B.V., Kingsfordweg 151, 1043GR, Amsterdam, The Netherlands
Pharmacodynamic properties
Pharmacotherapeutic group: Antihemorrhagics, other systemic hemostatics
ATC code: B02BX07
Mechanism of action
Lusutrombopag is an orally active TPO receptor agonist. Lusutrombopag acts on the haematopoietic stem cells and on the transmembrane domain of human TPO receptors expressed in megakaryocytes, to stimulate the megakaryocyte to proliferate and differentiate via the similar signal transduction pathway for up-regulating production used by endogenous TPO, thus leading to thrombocytopoiesis.
Clinical efficacy and safety
Two Phase 3, randomised, double-blind, placebo-controlled studies were conducted to evaluate lusutrombopag versus placebo in thrombocytopenic (platelet count <50,000/μL) subjects with chronic liver disease (Child-Pugh class A and B), undergoing elective invasive procedures (excluding laparotomy, thoracotomy, craniotomy, open-heart surgery, organ resection, or partial organ resection) in Japan (M0631 (L-PLUS 1)) and multiple countries (M0634 (L-PLUS 2)). Subjects were randomised to either of 3 mg of lusutrombopag or placebo in a 1:1 ratio. Randomisation was stratified by platelet count at screening/baseline and primary invasive procedure. Study drug was administered orally for up to 7 days. On Day 5 to Day 7, the platelet count was measured before administration of study drug. Administration of study drug was stopped if the platelet count was ≥50,000/μL together with an increase of ≥20,000/μL from baseline.
Invasive procedure was performed between Days 9 and 14.
In Study M0631, 96 subjects received lusutrombopag or placebo once daily: 48 subjects in the lusutrombopag group and 48 subjects in the placebo group. Eight lusutrombopag-treated subjects and 2 placebo-treated subjects received less than 7 days of treatment as they met the criterion for a responder prior to Day 7. Among the 48 subjects in the lusutrombopag group, 40 subjects received lusutrombopag for 7 days, 4 subjects for 6 days, 1 subject for 5 days, and 3 subjects for 4 days. Among the 48 subjects in the placebo group, 46 were treated for 7 days and 2 were treated for 4 days.
In Study M0634, 215 subjects were randomised in the study: 108 in the lusutrombopag 3 mg group and 107 in the placebo group. One subject in the lusutrombopag group withdrew from the study prior to administration of study drug. In the lusutrombopag group, 73/107 subjects (68.2%) received the study drug for 7 days. Of the remaining subjects in the lusutrombopag group, 15, 8, and 11 subjects received study drug for 4, 5, and 6 days, respectively. In the placebo group, 94/107 subjects (87.9%) received the study drug for 7 days. Of the remaining subjects in the placebo group, 5, 4, and 4 subjects received study drug for 4, 5, and 6 days, respectively.
The primary endpoint in Study M0631 was the proportion of subjects who required no platelet transfusion (i.e. achieved platelet count >50,000/μL) before the primary invasive procedure. The primary endpoint in Study M0634 was the proportion of subjects who required no platelet transfusion (i.e. achieved platelet count >50,000/μL) before the primary invasive procedure and no rescue therapy for bleeding from randomisation through 7 days after the primary invasive procedure.
In order to allow an overall comparison of the results across Studies M0631 and M0634, as presented in Table 2 to Table 5, data from study M0631 was reanalysed according to the primary endpoint for study M0634. The proportion of subjects who required no platelet transfusion prior to the primary invasive procedure and no rescue therapy for bleeding from randomisation through 7 days after the primary invasive procedure was statistically significantly greater in the lusutrombopag group compared with placebo group for the individual study and pooled analyses (Table 2).
Table 2. Proportion of subjects who required no platelet transfusion and no rescue therapy:
| Study M0631 | Study M0634 | Overall | ||||
|---|---|---|---|---|---|---|
| LUSU 3 mg N=49 | Placebo N=48 | LUSU 3 mg N=108 | Placebo N=107 | LUSU 3 mg N=157 | Placebo N=155 | |
| Proportion of subjects [a] (number of subjects) | 75.5% (37) | 12.5% (6) | 64.8% (70) | 29.0% (31) | 68.2% (107) | 23.9% (37) |
| Comparison with placebo [b]: Difference of proportion (95% CI) | 61.8 (46.4. 77.2) | 36.6 (24.6. 48.5) | 44.4 (34.9. 54.0) | |||
| P value | <0.0001 | <0.0001 | <0.0001 | |||
LUSU = lusutrombopag
[a] Proportion of subjects who required no platelet transfusion prior to the primary invasive procedure and no rescue therapy (including platelet transfusion) for bleeding from randomisation through 7 days after the primary invasive procedure. In addition to subjects who received platelet transfusion, subjects who did not receive an invasive procedure regardless of the reason were considered as receiving platelet transfusion.
[b] Cochran-Mantel-Haenszel test with baseline platelet count as stratum. In the analysis for pooled data, study was added as a stratum. The p value and confidence interval were calculated using the Wald method.
The key secondary endpoints in Studies M0631 and M0634 were
Proportion of subjects who required no platelet transfusion during the study (Day 1 through Day 35)
The proportion of subjects who required no platelet transfusion during the study was significantly greater in the lusutrombopag groups in the individual studies and the pooled (Studies M0631 and M0634) lusutrombopag group compared with placebo (Table 3).
Table 3. Proportion of Subjects who required no platelet transfusion during the study (Day 1 through Day 35):
| Study M0631 | Study M0634 | Overall | ||||
|---|---|---|---|---|---|---|
| LUSU 3 mg N=49 | Placebo N=48 | LUSU 3 mg N=108 | Placebo N=107 | LUSU 3 mg N=157 | Placebo N=155 | |
| Proportion of subjects [a] (number of subjects) | 77.6% (38) | 12.5% (6) | 63.0% (68) | 29.0% (31) | 67.5% (106) | 23.9% (37) |
| Comparison with placebo [b]: Difference of proportion (95% CI) | 63.8 (48.7. 78.9) | 34.7 (22.6. 46.8) | 43.8 (34.2. 53.4) | |||
| P value | <0.0001 | <0.0001 | <0.0001 | |||
[a] Proportion of subjects who required no platelet transfusion during the study (i.e., from Day 1 through Day 35). In addition to subjects who received platelet transfusion, subjects who did not receive an invasive procedure regardless of the re son were considered as receiving platelet transfusion.
[b] Cochran-Mantel-Haenszel test with baseline platelet count as stratum. In the analysis for pooled data, study was added as a stratum. The p value and confidence interval were calculated using the Wald method.
Proportion of responders
The proportion of subjects who met the responder criterion (defined as platelet count increase to ≥50,000/μL with an increase of ≥20,000/μL from baseline) during the study was significantly greater in the lusutrombopag groups in the individual studies and the pooled (Studies M0631 and M0634) lusutrombopag group compared with placebo (Table 4).
Table 4. Proportion of responders:
| Study M0631 | Study M0634 | Overall | ||||
|---|---|---|---|---|---|---|
| LUSU 3 mg N=49 | Placebo N=48 | LUSU 3 mg N=108 | Placebo N=107 | LUSU 3 mg N=157 | Placebo N=155 | |
| Proportion of subjects [a] (number of subjects) | 75.5% (37) | 6.3% (3) | 64.8% (70) | 13.1% (14) | 68.2% (107) | 11.0% (17) |
| Comparison with placebo [b]: Difference of proportion (95% CI) | 68.4 (54.4. 82.3) | 51.7 (41.1. 62.4) | 56.9 (48.4. 65.4) | |||
| P value | <0.0001 | <0.0001 | <0.0001 | |||
[a] A responder was defined as a subject who achieved a platelet count of ≥50,000/μL with an increase of ≥20,000/μL from baseline. A subject was considered a nonresponder if the subject met the responder criterion only after platelet transfusion.
[b] Cochran-Mantel-Haenszel test with baseline platelet count as stratum. In the analysis for pooled data, study was added as a stratum. The p value and confidence interval were calculated using the Wald method.
Duration of the increase in platelet count to ≥50,000/μL
The duration of the increase in platelet count to ≥50,000/μL in Studies M0631 and M0634 and the pooled (Studies M0631 and M0634) lusutrombopag group was significantly greater than compared with placebo (Table 5).
Table 5. Duration of the increase in platelet count to ≥50,000/μL:
| Study M0631 | Study M0634 | Overall | ||||
|---|---|---|---|---|---|---|
| LUSU 3 mg N=49 | Placebo N=48 | LUSU 3 mg N=108 | Placebo N=107 | LUSU 3 mg N=157 | Placebo N=155 | |
| Total | ||||||
| n | 48 | 48 | 107 | 107 | 155 | 155 |
| Median (days) | 21.1 | 3.4 | 15.1 | 1.0 | 17.3 | 1.8 |
| (Q1. Q3) | (13.7. 25.5) | (0.0. 11.3) | (6.6. 23.9) | (0.0. 9.2) | (9.7. 24.4) | (0.0. 9.5) |
| P value [a] | 0.0197 | 0.0002 | <0.0001 | |||
LUSU = lusutrombopag; Q1 = 25th percentile; Q3 = 75th percentile
[a] P-value was calculated by the van Elteren test with platelet transfusion status as stratum. In the analysis for pooled data, study was added as a stratum.
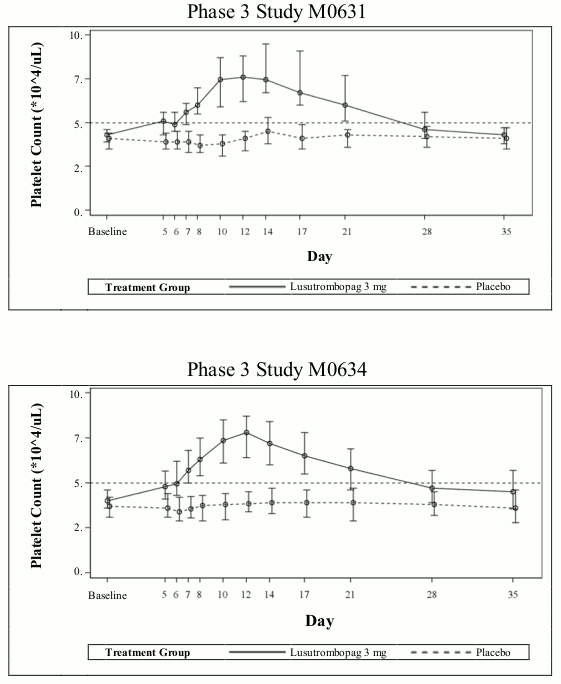
Time course of platelet count
The mean (range) maximum platelet count in subjects without platelet transfusion in the lusutrombopag group in Studies M0631 and M0634 was 90,200 (59,000 to 145,000)/μL and 86,900 (25,000 to 219,000)/μL, respectively; and the median (range) time to reach the maximum platelet count was 14.0 (6 to 28) days and 12.0 (5 to 35) days, respectively, and platelet count is expected to decrease thereafter. The time course of platelet counts in lusutrombopag-treated subjects without platelet transfusion and placebo-treated subjects with platelet transfusion in Studies M0631 and M0634 is presented in Figure 1.
Figure 1. Time course profiles of platelet count in the Phase 3 studies in thrombocytopenic patients with chronic liver disease (lusutrombopag-treated subjects without platelet transfusion and placebo-treated subjects with platelet transfusion):
Patients with severe hepatic impairment
In Study M0634, 3 subjects with Child-Pugh class C liver disease were erroneously enrolled (all in the lusutrombopag group). All 3 received 7 days of treatment with lusutrombopag. This limited data suggested no abnormal pattern of platelet count rise in this subpopulation.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with Mulpleo in all subsets of the paediatric population for thrombocytopenia secondary to liver disease (see section 4.2 for information on paediatric use).
Pharmacokinetic properties
Absorption
Lusutrombopag is absorbed with a peak concentration occurring 6 to 8 hours after oral administration. The accumulation ratios of the Cmax and the AUC are approximately 2 at once-daily multiple doses and the steady-state of the plasma concentration of lusutrombopag appear to be achieved after Day 5. The pharmacokinetics of lusutrombopag was similar in both healthy subjects and the chronic liver disease population. The pharmacokinetic parameters in patients with chronic liver disease are shown in Table 6.
Table 6. Pharmacokinetic parameters of lusutrombopag after 3 mg dose once daily in thrombocytopenic patients with chronic liver disease (Study M0634):
| Cmax (ng/mL) | Tmax (hr) | AUC0-τ (ng·hr/mL) | CL/F (L/hr) |
|---|---|---|---|
| 157 (34.7) | 5.95 (2.03, 7.85) | 2737 (36.1) | 1.10 (36.1) |
n=9.
Geometric mean (%CV) other than for Tmax, which is median (range).
Food interaction
Neither food (including high-fat and high-calorie diet) nor co-administration with calcium has a clinically meaningful effect on the pharmacokinetics of lusutrombopag.
Distribution
Human plasma protein binding ratio is ≥99.9%. The mean (% coefficient of variation) apparent volume of distribution during the terminal phase of lusutrombopag in healthy adult subjects (n=16) was 39.5 L (23.5%).
In rats, results indicated that lusutrombopag and its metabolites transfer to fetus via placenta.
Biotransformation
Lusutrombopag is a substrate of P-gp and BCRP, but is not a substrate of OATP1B1, OATP1B3 or OCT1. In the human mass balance study using [14C]-lusutrombopag, unchanged lusutrombopag (97% of radioactivity in plasma) was the major circulating component, and the metabolites, such as deshexyl, β-oxidated carboxylic acid, taurine conjugate of β-oxidated carboxylic acid, and acyl-glucuronide, were detected with less than 2.6% of radioactivity in plasma. In faeces, the components of radioactivity were unchanged lusutrombopag (16% of administered radioactivity) and β-oxidation-related metabolites (35% of administered radioactivity), suggesting that lusutrombopag is metabolised by ω-oxidation first, and subsequently metabolised by β-oxidation of O-hexyl side chain. In vitro studies revealed that CYP4 enzymes including CYP4A11 and partially CYP3A4 enzyme contributed to ω-oxidation to form 6-hydroxylated lusutrombopag. Drug interactions via inhibition and induction of any CYP4A enzymes have not been reported in clinical use. Therefore, inducers and inhibitors of CYP4A enzymes including CYP4A11 are unlikely to affect the pharmacokinetics of lusutrombopag. Lusutrombopag has low potential to inhibit CYP enzymes (CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4/5), and to induce both CYP enzymes (CYP1A2, 2C9, and 3A4) and UGT enzymes (UGT1A2, 1A6, and 2B7). Lusutrombopag has also low potential to inhibit P-gp, BCRP, OATP1B1, OATP1B3, OCT1, OCT2, OAT1, OAT3, MATE1, MATE2-K, and BSEP. Lusutrombopag is not considered to affect the pharmacokinetics of co-administered medicinal products that are substrates of these enzymes or transporters.
Elimination
Lusutrombopag was excreted mainly via faecal route in humans (approximately 83% into faeces and 1% into urine). Geometric mean of t1/2 (% coefficient of variation), was 38.3 hours (18.7%) after multiple oral dose of 3 mg lusutrombopag.
Linearity/non-linearity
Both Cmax and AUC for lusutrombopag increase dose-proportionally over dose range of multiple oral dose of 0.25 to 4 mg once daily in patients with chronic liver disease.
Pharmacokinetics in subpopulations
Age, gender and race
A population pharmacokinetic analysis using plasma lusutrombopag concentrations from clinical studies with lusutrombopag did not identify a clinically meaningful effect of age, gender or race on the pharmacokinetics of lusutrombopag.
Paediatric population
No pharmacokinetic data have been obtained in children.
Renal impairment
Lusutrombopag is rarely excreted into urine (approximately 1%). A population pharmacokinetic analysis using plasma lusutrombopag concentrations from clinical studies with lusutrombopag did not identify a clinically meaningful effect of renal function on the pharmacokinetics of lusutrombopag.
Hepatic impairment
Mild and moderate hepatic impairment (mild, Child-Pugh class A; moderate, Child-Pugh class B) is expected to have little effect on the pharmacokinetics of lusutrombopag. The differences in pharmacokinetics of a single 0.75 mg dose of lusutrombopag were relatively small in both subjects with mild hepatic impairment and subjects with moderate hepatic impairment, compared with the healthy matched control group. Ratios of AUC relative to the healthy matched control group were 1.05 in subjects with mild hepatic impairment and 1.20 in subjects with moderate hepatic impairment.
The ranges of observed Cmax and AUC0-τ overlapped among the patients with Child-Pugh class A, B, and C. Cmax and AUC0-τ of all patients with Child-Pugh class C did not exceed the maximum values from Child-Pugh class A and class B. Due to the limited information available, lusutrombopag should not be used in Child-Pugh class C patients unless the expected benefit outweighs the expected risks.
Preclinical safety data
Lusutrombopag does not stimulate platelet production in the species used for toxicological testing because of unique human TPO receptor specificity. Thus, the data from the toxicology program in these animals do not present potential adverse effects related to exaggerated pharmacology in humans.
Effects in non-clinical studies were observed only at exposures considered sufficiently in excess of the maximum human exposure indicating little relevance to clinical use.
In rats, lusutrombopag and its metabolites are excreted in milk, and the concentrations in milk decreased as with those in plasma.
Repeated toxicity
The principal toxicity findings associated with lusutrombopag administration included prolongation of PT and APTT (rats), increased activities of plasma ALT and AST (rats and dogs), adrenal toxicity (rats and dogs), skin and forestomach lesions (rats) and renal toxicity (rats).
High dose (10 mg/kg/day) and long-term treatment (8 weeks) of lusutrombopag has a potential risk of fibrosis in the bone marrow via human TPO receptor based on the results of study in TPOR-Ki/Shi mice with chimeric human transmembrane domain TPO receptor knocked-in to the mouse TPO receptor.
Carcinogenesis
Lusutrombopag was not carcinogenic to mice at doses up to 20 mg/kg/day in males and females (a dose at least 45 times the human clinical exposures in adults based on AUC), or rats at doses up to 20 mg/kg/day in males and 2 mg/kg/day in females (a dose 49 and 30 times, respectively, the human clinical exposures in adults based on AUC).
Mutagenesis
Lusutrombopag was not genotoxic when tested in a bacterial reverse mutation test, a chromosomal aberration test with cultured Chinese hamster lung cells, or an in vivo micronucleus test with mouse bone marrow cells.
Fertility
Lusutrombopag did not affect male and female fertility and early embryo development in rats at doses up to 100 mg/kg/day (176 and 252 times respectively, the human clinical exposures in adults based on AUC).
Embryo-foetal development
Lusutrombopag showed no teratogenicity in rats and rabbits at up to 80 mg/kg/day and 1000 mg/kg/day respectively. No effects on foetal viability embryo-foetal development were noted in rabbits at doses up to 1000 mg/kg/day (161 times the human clinical exposures in adults based on AUC). In rats, there were adverse effects of lusutrombopag on foetal intrauterine growth and skeletal morphology as follows: a suppression of foetal intrauterine growth (low foetal body weight and a decrease in the number of ossified sternebrae) at 80 mg/kg/day, and an high incidence of short cervical supernumerary ribs at 40 mg/kg/day or more, and an high incidence of short thoracolumbar supernumerary rib at 4 mg/kg/day or more. A suppression of foetal intrauterine growth as well as cervical ribs occurred at doses (40 mg/kg/day or more), showing maternal toxicity. Meanwhile, the short thoracolumbar supernumerary ribs were observed at doses without maternal toxicity. The changes were also noted in F1 pups on postnatal day (PND) 4 at 12.5 mg/kg/day or more in the pre- and postnatal development study; however, F1 mature animals showed no full and short thoracolumbar supernumerary rib. On the basis of the results, the no observed adverse effect level (NOAEL) was estimated to be near 4 mg/kg/day in the embryo-foetal development study in rats (23 times the human clinical exposures in adults based on AUC).
Pre- and post-natal development
In the pre- and postnatal development study in rats at doses up to 40 mg/kg/day, there were adverse effects of lusutrombopag on postnatal development at 40 mg/kg/day as follow: prolongation of gestation period in dams, low viability before weaning, delayed postnatal growth such as delayed negative geotaxis or delayed eyelid opening, low pup body weight, low female fertility index, a tendency to low number of corpora lutea or implantations, and a tendency to increased pre-implantation loss rate and an abnormal clinical sign such as prominent annular rings on tail after weaning. There were no effects on pregnancy, parturition, lactation in F0 dams and postnatal development in F1 pups at doses up to 12.5 mg/kg/day (89 times the human clinical exposures in adults based on AUC).
Phototoxicity
Lusutrombopag has no phototoxic potential in the skin phototoxicity study in hairless mice at doses up to 500 mg/kg (96.3 μg/mL) (613 times the human clinical exposures in adults based on Cmax [0.157 μg/mL]).
Environmental Risk Assessment (ERA)
Environmental risk assessment studies have shown that lusutrombopag has the potential to be very persistent, very bioaccumulative and toxic to the environment.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.