Source: European Medicines Agency (EU) Revision Year: 2019 Publisher: Astellas Pharma Europe B.V., Sylviusweg 62, 2333 BE Leiden, Netherlands
Pharmacotherapeutic group: Antimycotics for systemic use, other antimycotics for systemic use
ATC code: J02AX05
Micafungin non-competitively inhibits the synthesis of 1,3-β-D-glucan, an essential component of the fungal cell wall. 1,3-β-D-glucan is not present in mammalian cells.
Micafungin exhibits fungicidal activity against most Candida species and prominently inhibits actively growing hyphae of Aspergillus species.
In animals models of candidiasis, a correlation was observed between exposure of micafungin divided by MIC (AUC/MIC) and efficacy defined as the ratio required to prevent progressive fungal growth. A ratio of ~2400 and ~1300 was required for C. albicans and C. glabrata, respectively, in these models. At the recommended therapeutic dosage of Mycamine, these ratios are achievable for the wild-type distribution of Candida spp.
As for all antimicrobial agents, cases of reduced susceptibility and resistance have been reported and cross-resistance with other echinocandins cannot be excluded. Reduced susceptibility to echinocandins has been associated with mutations in the Fks1 and Fks2 genes coding for a major subunit of glucan synthase.
EUCAST breakpoints:
| Candida species | MIC breakpoint (mg/L) | |
|---|---|---|
| ≤S (Susceptible) | >R (Resistant) | |
| Candida albicans | 0.016 | 0.016 |
| Candida glabrata | 0.03 | 0.03 |
| Candida parapsilosis | 0.002 | 2 |
| Candida tropicalis1 | Insufficient evidence | |
| Candida krusei1 | Insufficient evidence | |
| Candida guilliermondii1 | Insufficient evidence | |
| Other Candida spp. | Insufficient evidence | |
1 MICs for C. tropicalis are 1-2 two-fold dilution steps higher than for C albicans and C. glabrata. In the clinical study, successful outcome was numerically slightly lower for C. tropicalis than for C. albicans at both dosages (100 and 150 mg daily). However, the difference was not significant and whether it translates into a relevant clinical difference is unknown. MICs for C. krusei are approximately 3 two-fold dilution steps higher than those for C. albicans and, similarly, those for C. guilliermondii are approximately 8 two-fold dilutions higher. In addition, only a small number of cases involved these species in the clinical trials. This means there is insufficient evidence to indicate whether the wild-type population of these pathogens can be considered susceptible to micafungin.
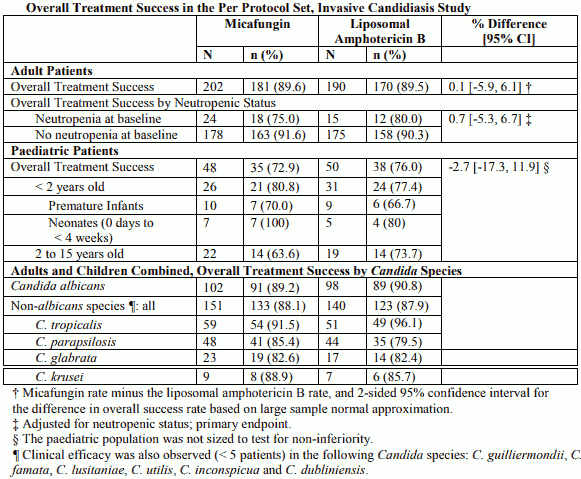
Candidaemia and Invasive Candidiasis: Micafungin (100 mg/day or 2 mg/kg/day) was as effective as and better tolerated than liposomal amphotericin B (3 mg/kg) as first-line treatment of candidaemia and invasive candidiasis in a randomised, double-blind, multinational non-inferiority study. Micafungin and liposomal amphotericin B were received for a median duration of 15 days (range, 4 to 42 days in adults; 12 to 42 days in children).
Non-inferiority was proven for adult patients, and similar findings were demonstrated for the paediatric subpopulations (including neonates and premature infants). Efficacy findings were consistent, independent of the infective Candida species, primary site of infection and neutropenic status (see Table). Micafungin demonstrated a smaller mean peak decrease in estimated glomerular filtration rate during treatment (p<0.001) and a lower incidence of infusion-related reactions (p=0.001) than liposomal amphotericin B.
Oesophageal Candidiasis: In a randomised, double-blind study of micafungin versus fluconazole in the first-line treatment of oesophageal candidiasis, 518 patients received at least a single dose of study drug. The median treatment duration was 14 days and the median average daily dose was 150 mg for micafungin (N=260) and 200 mg for fluconazole (N=258). An endoscopic grade of 0 (endoscopic cure) at the end of treatment was observed for 87.7% (228/260) and 88.0% (227/258) of patients in the micafungin and fluconazole groups, respectively (95% CI for difference: [-5.9%, 5.3%]). The lower limit of the 95% CI was above the predefined non-inferiority margin of -10%, proving non-inferiority. The nature and incidence of adverse events were similar between treatment groups.
Prophylaxis: Micafungin was more effective than fluconazole in preventing invasive fungal infections in a population of patients at high risk of developing a systemic fungal infection (patients undergoing haematopoietic stem cell transplantation [HSCT] in a randomised, double-blind, multicentre study). Treatment success was defined as the absence of a proven, probable, or suspected systemic fungal infection through the end of therapy and absence of a proven or probable systemic fungal infection through the end of study. Most patients (97%, N=882) had neutropenia at baseline (< 200 neutrophils/µL). Neutropenia persisted for a median of 13 days. There was a fixed daily dose of 50 mg (1.0 mg/kg) for micafungin and 400 mg (8 mg/kg) for fluconazole. The mean period of treatment was 19 days for micafungin and 18 days for fluconazole in the adult population (N=798) and 23 days for both treatment arms in the paediatric population (N=84).
The rate of treatment success was statistically significantly higher for micafungin than fluconazole (1.6% versus 2.4% breakthrough infections). Breakthrough Aspergillus infections were observed in 1 versus 7 patients, and proven or probable breakthrough Candida infections were observed in 4 versus 2 patients in the micafungin and fluconazole groups, respectively. Other breakthrough infections were caused by Fusarium (1 and 2 patients, respectively) and Zygomycetes (1 and 0 patients, respectively). The nature and incidence of adverse reactions were similar between treatment groups.
Pharmacokinetics are linear over the daily dose range of 12.5 mg to 200 mg and 3 mg/kg to 8 mg/kg. There is no evidence of systemic accumulation with repeated administration and steady-state is generally reached within 4 to 5 days.
Following intravenous administration concentrations of micafungin show a biexponential decline. The drug is rapidly distributed into tissues. In systemic circulation, micafungin is highly bound to plasma protein (>99%), primarily to albumin. Binding to albumin is independent of micafungin concentration (10-100 µg/ml). The volume of distribution at steady state (Vss) was approximately 18-19 litres.
Unchanged micafungin is the principal circulating compound in systemic circulation. Micafungin has been shown to be metabolised to several compounds; of these M-1 (catechol form), M-2 (methoxy form of M-1) and M-5 (hydroxylation at the side chain) of micafungin have been detected in systemic circulation. Exposure to these metabolites is low and metabolites do not contribute to the overall efficacy of micafungin.
Even though micafungin is a substrate for CYP3A in vitro, hydroxylation by CYP3A is not a major pathway for micafungin metabolism in vivo.
The mean terminal half-life is approximately 10-17 hours and stays consistent across doses up to 8 mg/kg and after single and repeated administration. Total clearance was 0.15-0.3 ml/min/kg in healthy subjects and adult patients and is independent of dose after single and repeated administration. Following a single intravenous dose of 14C-micafungin (25 mg) to healthy volunteers, 11.6% of the radioactivity was recovered in the urine and 71.0% in the faeces over 28 days. These data indicate that elimination of micafungin is primarily non-renal. In plasma, metabolites M-1 and M-2 were detected only at trace concentrations and metabolite M-5, the more abundant metabolite, accounted for a total of 6.5% relative to parent compound.
In paediatric patients AUC values were dose proportional over the dose range of 0.5-4 mg/kg. Clearance was influenced by weight, with mean values of weight-adjusted clearance 1.35 times higher in the younger children (4 months to 5 years) and 1.14 times higher in paediatric patients aged 6 to 11 years. Older children (12-16 years) had mean clearance values similar to those determined in adult patients. Mean weight-adjusted clearance in children less than 4 months of age is approximately 2.6-fold greater than older children (12-16 years) and 2.3-fold greater than in adults.
PK/PD bridging study demonstrated dose-dependent penetration of micafungin into CNS with the minimum AUC of 170 µg*hr/L required to achieve maximum eradication of fungal burden in the CNS tissues. Population PK modeling demonstrated that a dose of 10 mg/kg in children less than 4 month of age would be sufficient to achieve the target exposure for the treatment of CNS Candida infections.
When administered as a single 1-hour infusion of 50 mg the pharmacokinetics of micafungin in the elderly (aged 66-78 years) were similar to those in young (20-24 years) subjects. No dose adjustment is necessary for the elderly.
In a study performed in patients with moderate hepatic impairment (Child-Pugh score 7-9), (n=8), the pharmacokinetics of micafungin did not significantly differ from those in healthy subjects (n=8). Therefore, no dose adjustment is necessary for patients with mild to moderate hepatic impairment. In a study performed in patients with severe hepatic impairment (ChildPugh score 10-12) (n=8), lower plasma concentrations of micafungin and higher plasma concentrations of the hydroxide metabolite (M-5) were seen compared to healthy subjects (n=8). These data are insufficient to support a dosing recommendation in patients with severe hepatic impairment.
Severe renal impairment (Glomerular Filtration Rate [GFR] <30 ml/min) did not significantly affect the pharmacokinetics of micafungin. No dose adjustment is necessary for patients with renal impairment.
Gender and race (Caucasian, Black and Oriental) did not significantly influence the pharmacokinetic parameters of micafungin. No dose adjustment of micafungin is required based on gender or race.
The development of foci of altered hepatocytes (FAH) and hepatocellular tumours in rats was dependent on both dose and duration of micafungin treatment. FAH recorded after treatment for 13 weeks or longer persisted after a 13-week withdrawal period and developed into hepatocellular tumours following a treatment free period which covered the life span of rats. No standard carcinogenicity studies have been conducted but the development of FAH was assessed in female rats after up to 20 and 18 months after cessation of a 3 and 6 month treatment, respectively. In both studies increased incidences/numbers of hepatocellular tumours were observed after the 18 and 20 month treatment free period in the high dose group of 32 mg/kg/day as well as in a lower dose group (although not statistically significant). The plasma exposure at the assumed threshold for tumour development in rats (i.e. the dose where no FAH and liver tumours were detected) was in the same range as the clinical exposure. The relevance of the hepatocarcinogenic potential of micafungin for the human therapeutic use is not known.
The toxicology of micafungin following repeated intravenous dosing in rats and/or dogs showed adverse responses in liver, urinary tract, red blood cells, and male reproductive organs. The exposure levels at which these effects did not occur (NOAEL) were in the same range as the clinical exposure or lower. Consequently, the occurrence of these adverse responses may be expected in human clinical use of micafungin.
In standard safety pharmacology tests, cardiovascular and histamine releasing effects of micafungin were evident and appeared to be time above threshold dependent. Prolongation of infusion time reducing the plasma concentration peak appeared to reduce these effects.
In repeated dose toxicity studies in rat signs of hepatotoxicity consisted of increased liver enzymes and degenerative changes of hepatocytes which were accompanied by signs of compensatory regeneration. In dog, liver effects consisted of increased weight and centrilobular hypertrophy, no degenerative changes of hepatocytes were observed.
In rats, vacuolation of the renal pelvic epithelium as well as vacuolation and thickening (hyperplasia) of the bladder epithelium were observed in 26-week repeat dose studies. In a second 26-week study hyperplasia of transitional cells in the urinary bladder occurred with a much lower incidence. These findings showed reversibility over a follow-up period of 18 months. The duration of micafungin dosing in these rat studies (6 months) exceeds the usual duration of micafungin dosing in patients (see section 5.1).
Micafungin haemolysed rabbit blood in vitro. In rats, signs of haemolytic anaemia were observed after repeated bolus injection of micafungin. In repeat dose studies in dogs, haemolytic anaemia was not observed.
In reproductive and developmental toxicity studies, reduced birth weight of the pups was noted. One abortion occurred in rabbits at 32 mg/kg/day. Male rats treated intravenously for 9 weeks showed vacuolation of the epididymal ductal epithelial cells, increased epididymis weights and reduced number of sperm cells (by 15%), however, in studies of 13 and 26 weeks duration these changes did not occur. In adult dogs, atrophy of seminiferous tubules with vacuolation of the seminiferous epithelium and decreased sperm in the epididymides were noted after prolonged treatment (39 weeks) but not after 13 weeks of treatment. In juvenile dogs, 39 weeks treatment did not induce lesions in the testis and epididymides in a dose dependent manner at the end of treatment but after a treatment free period of 13 weeks a dose dependent increase in these lesions were noted in the treated recovery groups. No impairment of male or female fertility was observed in the fertility and early embryonic development study in rats.
Micafungin was not mutagenic or clastogenic when evaluated in a standard battery of in vitro and in vivo tests, including an in vitro study on unscheduled DNA synthesis using rat hepatocytes.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.